1Division of Endocrinology and Metabolism, Department of Medicine, Kurume University School of Medicine, Kurume, 2Division of Surgery, Seishinkai Inoue Hospital, Alumni of the 1st Department of Surgery, Kyushu University and Japanese Society for the Medical History, Itoshima, Fukuoka, 3Kuma Hospital, Center for Excellence in Thyroid Care, Chuo-ku, Kobe, Japan
Hashimoto’s thyroiditis, Chronic thyroiditis, IgG4 thyroiditis, History, Hakaru Hashimoto
1. Introduction
Hashimoto’s thyroiditis (HT), a common thyroid disease, is now recognized as an autoimmune thyroid disorder (AITD). However, surprisingly little is known about its discoverer, Hakaru Hashimoto, who first described the disease in 1912.1 He presented four patients with a chronic thyroid disorder, which he termed struma lymphomatosa, characterized by diffuse lymphocytic infiltration with germinal centers, fibrosis, parenchymal atrophy and eosinophilic change in some thyroid follicular cells. In this review, we recount the discovery of HT by Hakaru Hashimoto, its identification as an AITD, current concepts concerning its etiology, pathology, clinical diagnosis and histological variants of HT and the outlook for this disease.
2. Discovery of Hashimoto’s thyroiditis
Dr. Hakaru Hashimoto (Figure 1), born 5 May 1881, was the third son of a prominent family who had practiced medicine for generations in Iga Ueno, Mie Prefecture, Japan.2-5 He was one of the first graduates to receive a bachelor’s degree from the Fukuoka Medical College of Kyoto Imperial University in 1907. Hakaru Hashimoto received tutelage and training from Professor Miyake of the Department of Surgery of Fukuoka Medical College. Professor Miyake also instructed Dr. Hashimoto on the technique of assessing excised thyroid glands microscopically. In 1912, at the age of 30, Hakaru Hashimoto reported a distinct new disease in ‘Archiv für Klinische Chirurgie”, a German journal of clinical surgery.1 His paper, which consisted of 30 pages and 5 illustrations and centered on histological changes in thyroid tissue, discussed the results of his examination of thyroid tissue samples taken from four women. Hakaru Hashimoto further explained in his report that the new pathological characteristics he had identified, namely infiltration of lymphoid and plasma cells, formation of lymphoid follicles with germinal centers, fibrosis, degenerated thyroid epithelial cells and leukocytes in the lumen, were histologically similar to those of Mikulicz’s disease. He expressed confidence that he had discovered a new disease, which he named “struma lymphomatosa”, emphasizing the lymphoid cell infiltration and formation of lymphoid follicles with germinal centers, neither of which had previously been reported.

Figure 1. Portrait of Dr. Hakaru Hashimoto at age 31.
After the publication of his paper, Dr. Hashimoto studied pathological science under Professor Kaufmann, a leading German pathologist at the Göttingen University, where he researched renal tuberculosis for two years. He returned to Japan at the outbreak of the First World War in 1914. In 1916, he took over the family practice in his hometown. Since he was a graduate of the Imperial University and had furthered his education abroad, his reputation quickly spread throughout the country. Throughout his professional life, he treated all his patients with equal devotion and concern. He died of typhoid fever on 9 January 1934 at the age of 52, unfortunately without receiving the recognition he merited for his discovery, which would later be called Hashimoto’s disease.
In spite of their different clinical manifestations, Hashimoto’s lymphomatous thyroiditis was not recognized as distinct from Riedel’s thyroiditis, a common disease at that time in Europe.4 Although Simmonds et al6 in 1913 and Heineke et al7 in 1914 published case reports similar to Hashimoto’s description, the German school asserted that Hashimoto’s struma lymphomatosa was an early phase of Riedel’s thyroiditis.8,9 Hashimoto’s struma lymphomatosa was then ignored and forgotten until 1931, when Allen Graham et al10 of Cleveland reported struma lymphomatosa as detailed by Hakaru Hashimoto and endorsed Hashimoto’s conclusion that it was a disease in its own right. Since then, this disease has been eponymously referred to as HT.
In 1956, Drs. Rose and Witebsky11,12 showed that immunization of rabbits with extracts of rabbit thyroid produced histological changes in the thyroid glands resembling those seen in HT. They also found anti-thyroglobulin antibodies in sera from these animals. In the same year, Deborah Doniach and colleagues13 of Middlesex Hospital, London, purified an anti-thyroglobulin antibody from the sera of patients with HT and asserted that patients with HT have an immunological reaction to human thyroglobulin. Doniach also proposed that Hashimoto’s struma is an autoimmune disease of the thyroid gland. Following these discoveries, the concept of organ-specific autoimmune disease was established and HT recognized as one such disease. Doniach’s “letter to the editors” published in the Lancet in 1962 included a portrait of Hakaru Hashimoto that had been taken in Göttingen in 1912.14 From that moment on, Hakaru Hashimoto’s image and achievements became well known all over the world and the terms Hashimoto’s disease and HT increasingly appeared in textbooks.15 Since the discovery that Hashimoto’s disease is an autoimmune disorder, autoimmune abnormalities have been found in many other diseases. Today, a large number of diseases have been discovered to be autoimmune disorders and several thyroid-specific autoantibodies have been identified. Antibodies against thyroid peroxidase (TPOAb) are present in 10% of women in the general population.16 Painless thyroiditis and postpartum thyroiditis frequently develop in patients with HT.17,18
3. Hashimoto’s thyroiditis at present
3.1. Current concepts
Hashimoto’s thyroiditis is an organ-specific AITD characterized by diffuse goiter with lymphocytic infiltration and the presence of thyroid-specific autoantibodies. Today, it is one of the most widespread thyroid disorders and a cause of hypothyroidism in areas of the world where dietary iodine is sufficient. The incidence is 0.3–1.5 cases per 1000 population per year. The prevalence of positive antibody tests in women is greater than 10% and of clinical disease at least 2%.19 Men have one tenth of this prevalence.
3.2. Etiology
Although the exact etiology is not as yet known, HT is thought to arise from an interaction between genetic susceptibility factors, epigenetic effects and various environmental triggers (e.g. iodine, infection).20
3.2.1 Genetic susceptibility
Twin studies showed epidemiological evidence for genetic susceptibility to HT. The concordance rate for monozygotic twins was 55% vs. 0% for dizygotic twins in HT.21 The concordance rates for thyroid autoantibodies were also higher in monozygotic twins than in dizygotic twins (80% vs. 40%). To date, several loci have been associated with Hashimoto’s disease, such as HLA-DR, immune-regulatory genes (CD40, CTLA-4, PTPN22, FOXP3 and CD25) and thyroid-specific genes (thyroglobulin and thyroid-stimulating hormone [TSH] receptor). HLA-DRb1 Tyr26, Gln70, Lys71 and Arg74 may cause structural changes in pocket 4 and therefore influence binding of certain thyroid-derived pathogenic peptides.22 Tyr30 may also cause structural changes in pocket 6. These structural changes can affect the selectivity and binding of pathogenic peptides to these pockets, influencing the risk of Hashimoto’s disease. A -1623 A/G single-nucleotide polymorphism at thyroglobulin (Tg) promoter (rs180195) defines a cis-regulatory element that may influence binding of nuclear transcription factors such as interferon regulatory factor-1 protein. Increased production of interferon (IFN)g in viral infection may increase expression of Tg and lead to activation of T-cell responses in genetically susceptible individuals.23 A C/T polymorphism in the Kozak sequence of the cluster of differentiation (CD) 40 gene has significant effects on CD40 on antigen-presenting cells, including B cells, and therefore influences B cell proliferation, immunoglobulin class switching, antibody secretion and generation of memory cells.24 Cytotoxic T lymphocyte-associated antigen-4 (CTLA4) and protein tyrosine phosphatase-22 (PTPN22) are major negative regulators of T cell-mediated immune functions. Polymorphisms of the CTLA4 and PTPN22 genes and Hashimoto’s thyroiditis are linked.25 However, the mechanisms by which they confer susceptibility to thyroid autoimmunity are unknown. In the TSH receptor-immunized murine model of Graves’ disease, regulatory T cells (Treg) depletion induces thyroid lymphocytic infiltration with transient or permanent hypothyroidism.26 This suggests that Tregs play a role in the natural progression of hyperthyroid Graves’ disease to Hashimoto’s disease and hypothyroidism in humans.27
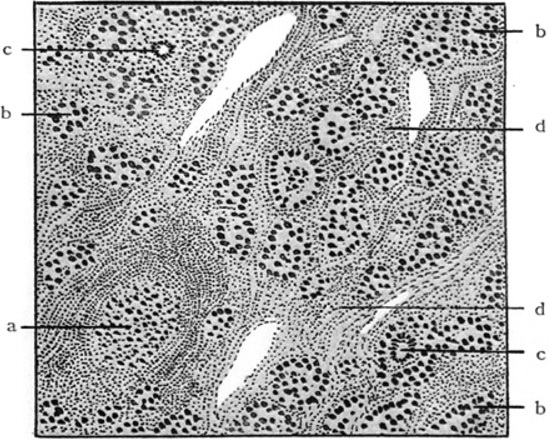
Figure 2. Histopathologic features of struma lymphomatosa as depicted in the original paper1 a: Lymphoid follicle; b: Degenerated thyroid follicle; c: Giant cells; d: Hyperplastic interstitium with prominent round cell infiltration.
3.2.2 Environmental triggers
Even with identical twins the concordance rate was only about 50%, emphasizing that other important factors such as environmental factors play roles in the pathogenesis of HT. In genetically susceptible individuals, the environmental factors, such as infection, dietary factors (iodine), stress and pregnancy, may initiate autoimmunity in the thyroid.
Although excess intake of iodine directly blocks hormone synthesis or is directly toxic to thyrocytes, iodine can induce autoimmunity in the thyroid by increasing the immunogenicity of thyroglobulin molecules and/or releasing free oxygen radicals via the enzymatic reaction of thyroid peroxidase. The increased free radicals induce an elevated expression of intracellular adhesion molecule-1.28 However, the precise mechanism for iodine’s induction of autoimmunity in the thyroid is not fully elucidated.
The other environmental factors, such as smoking, hepatitis C virus infection or selenium deficiency, may increase the autoimmune reaction and subsequent inflammation in the thyroid.29, 30 More recently, it has been proposed that viral infection and innate receptors may play a role in the etiology of HT.31 However, the precise mechanisms of the development of HT or their roles in the pathogenesis of HT have not been fully elucidated. Future studies are required to study the molecular basis of the interaction between environmental factors and susceptibility genes.
3.3. Pathology
The thyroid gland is pale and rubbery to firm. The characteristic histopathologic changes of HT include diffuse lymphoplasmacytic infiltration, lymphoid follicle formation with germinal centers, a varying degree of fibrosis, parenchymal atrophy and the presence of large follicular cells with abundant granular eosinophilic cytoplasm, so-called Hürthle, oxyphilic or Askanazy cells (Figure 2).19 Foreign body giant cells and granulomas are not features of HT. Electromicroscopy shows deposits of immunoglobulin (Ig) G and Tg along the basement membranes of follicular cells. Both B and T cells infiltrate the thyroid. Most infiltrating T cells have alpha/beta T cell receptors. Increased expression of Th1-related cytokines such as IFNg, interleukin-2 and CD25 reportedly occurs in intrathyroidal T cells from patients with HT. The impairment of hormone synthesis is due to apoptotic destruction of thyroid cells.32 Increased expression of Fas on thyroid follicular cells by the surrounding cytokines and Fas ligands on T cells may induce this apoptosis, although other cell death pathways, such as complement-fixing cytotoxicity and thyroglobulin-specific T cell-mediated cytotoxicity, may also be involved.33
3.4. Clinical Diagnosis
Clinical diagnosis of HT is based on the presence of diffuse goiter, anti-thyroid peroxidase Abs, anti-Tg Abs or lymphocytic infiltration on cytological examination. However, various clinical courses can reportedly occur, including euthyroidism with goiter, subclinical hypothyroidism with goiter, hypothyroidism, adolescent goiter, painless thyroiditis (silent thyroiditis), postpartum painless thyroiditis, alternating hypo-and hyperthyroidism and other accompanying autoimmune diseases. Painless thyroiditis is characterized by transient thyrotoxic phase with no anterior neck pain or tenderness, this latter condition passing through euthyroidism to hypothyroidism and, finally, returning to euthyroidism.34 Postpartum thyroiditis occurs within 6 months after delivery and is identical to painless thyroiditis. An immune rebound mechanism may mediate induction of painless thyroiditis and postpartum thyroiditis with transient autoimmune phenomena.34
3.5. Histological variants
Histopathologically HT is heterogenous. There are several histological variants, including fibrous35 and atrophic variants,35,36 Riedel’s thyroiditis,9,37,38 IgG4 thyroiditis.39-41
3.5.1. Fibrous variant
About 10-13% of all patients with HT exhibit this subtype. The normal thyroid parenchyma is destroyed and extensively replaced by cellular fibrous tissue and collagenous stroma. The fibrosis does not extend beyond the thyroid capsule. Clinically, a very firm goiter with a history of recent enlargement, marked hypothyroidism and a marked increase in anti-thyroglobulin antibody titer are characteristic of this variant. This condition can be mistakenly diagnosed as malignancy.
3.5.2. Fibrous atrophy variant
The gland is very small (1 to 6 g) and is characterized by abundant fibrous tissue, parenchymal atrophy and lymphoplasmacytic infiltration.32,33 This variant is clinically associated with myxedema and a high incidence of Tg Abs. In the blocking type of anti-TSH, receptor, antibodies are present in the sera of 10% of patients with atrophic thyroiditis.42
3.5.3. Riedel’s thyroiditis
Bernhard Riedel first recognized this disorder in 1896; he described two patients with hard goiters and tracheal compressive symptoms.9 Later, some experts suggested that RT is not a primary thyroid disease but rather a manifestation of the systemic disorder, multifocal fibrosclerosis.37 The inclusion of Riedel’s thyroiditis among autoimmune thyroid disorders is still debated. One third of RT cases have associated clinical findings of multifocal fibrosclerosis at the time of diagnosis. The incidence of RT was 0.06% in a series of 57,000 thyroidectomies at the Mayo Clinic.38 The histological criteria are as follows: (I) a fibro-inflammatory process involving all or a portion of the thyroid gland; (ii) fibrous extension beyond the thyroid capsule into adjacent anatomic structures; (iii) infiltrates of inflammatory cells without giant cells, lymphoid follicles, oncocytes or granulomas; (iv) evidence of occlusive phlebitis; and (v) absence of neoplasia.
3.5.4. IgG4 thyroiditis
Immunoglobulin G4 related disease (IgG4-RD) is now widely recognized as a multi-organ system disease characterized by tumefactive enlargement of affected organs and increased serum and tissue concentrations of IgG4.39 Very recently a unique subtype of HT, termed ‘IgG4 thyroiditis’, has been proposed.40 It is characterized by lymphoplasmacytic infiltration, fibrosis, increased IgG4-positive plasma cells in the thyroid and high serum IgG4 concentrations. More than 20 IgG4-positive plasma cells/high power field (HPF) and a ratio of >30% IgG4 positive/IgG positive plasma cells have been proposed as diagnostic criteria for IgG4 thyroiditis. IgG4 immunostaining, degree of fibrosis and invasive fibrosis beyond the thyroid capsule may be important aids to making the differential diagnosis. IgG4 thyroiditis is clinically associated with a lower female-to-male ratio, more rapid progression, subclinical hypothyroidism, diffuse low echogenicity and higher concentrations of circulating thyroid autoantibodies compared to those in non-IgG4 thyroiditis. IgG4 thyroiditis is clinically associated with IgG4-RD. Although fibrous variant and Riedel’s thyroiditis may belong to the spectrum of IgG4-RD,39,41 further studies are necessary to clarify this.
3.6. Hashimoto’s encephalopathy
In 1966 Brain et al. described a case of episodic encephalopathy associated with HT.43 Since then, many cases of corticosteroid-responsive encephalopathy associated with positive anti-thyroid antibodies have been reported and this type of encephalopathy is now termed Hashimoto’s encephalopathy.44 A recent study has proposed that human a-enolase is the responsible autoantigen in Hashimoto’s encephalopathy. Although detection of anti-a-enolase antibody is useful in diagnosis and management of Hashimoto’s encephalopathy, there is no direct evidence linking thyroid dysfunction or anti-thyroid antibodies with clinical features of the encephalopathy.45
3.7. Association of HT with papillary thyroid carcinoma and malignant lymphoma
In addition to the well-established relationship between HT and thyroid lymphoma,46 the risk of papillary thyroid carcinoma is also increased in patients with HT.47 These cancers seem no more aggressive than other papillary thyroid carcinomas.
4. Outlook for Hashimoto’s thyroiditis
Although significant progress has been made in our understanding of the contributions of genetic factors and environmental triggers to thyroid autoimmunity, the mechanisms by which gene variants interact with environmental factors to cause thyroid autoimmunity are still unclear. Recent advances in molecular technology will likely provide better understanding of the relationship between genetic factors and environmental factors in HT and new therapeutic approaches to HT. Many organ-specific autoimmune disorders are reportedly associated with HT. The new concept of IgG4 thyroiditis may help clinicians with the diagnosis and management of HT. Future studies on the mechanisms of the association between HT and other autoimmune disorders and/or thyroid malignancy are essential.
Acknowledgments
The authors gratefully acknowledge the invaluable assistance rendered by the late Dr. Kazuo Hashimoto, Dr. Hakaru Hashimoto’s son.
REFERENCES
1. Hashimoto H. 1912 Zur Kenntniss der lymphomatösen Veränderung der Schilddrüse (Struma lymphomatosa). Arch Klin Chir 97: 219-248.2. Hashimoto K, 2002 My father and his teacher. Endocr J 49: 389-391.
3. Amino N, Tada H, Hidaka Y, et al, 2002 Hashimoto’s disease and Dr. Hakaru Hashimoto. Endocr J 49: 393-397.
4. Sawin CT, 2002 The heritage of Dr. Hakaru Hashimoto (1881-1934). Endocr J 49: 399-403.
5. Satoh H, 2011 The history of Hashimoto disease: The establishment of a new diagnosis “Hashimoto disease” with a biography of Hakaru Hashimoto and a brief history of Fukuoka Medical College. In: Commemorative article of a centennial of Hashimoto disease in the Bulletin of the 1st Department of Surgery, Kyushu University, pp 9-21.
6. Simmonds M, 1913 Über lymphatische Herde in der Schilddrüse. Virch Arch Patholog Anat Physiol 211: 73-89.
7. Heineke, 1914 Die chronische Thyreoiditis. Deut Zeit Chir 129: 189-220.
8. Reist A, 1922 Über chronische Thyreoiditis. Frankf Zeit f Pathol 28: 141-200.
9. Riedel BM, 1896 Die chronische zur Bildung eisenharter Tumoren führende Entzündung der Schilddruese. Verh Ges Chir 25:101-105.
10. Graham A, 1931 Riedel’s struma in contrast to struma lymphomatosa (Hashimoto). West J Surg 39: 681-689.
11. Rose NR, Witebsky E, 1956 Studies on organ specificity: V. Changes in the thyroid glands of rabbits following active immunization with rabbit thyroid extracts. J Immunol 76: 417-427.
12. Witebsky E, Rose NR, Paine JR, et al, 1957 Thyroid-specific autoantibodies. Ann N Y Acad Sci 69: 669-677.
13. Campbell PN, Doniach D, Hudson RV, et al, 1956 Auto-antibodies in Hashimoto’s disease (lymphadenoid goitre). Lancet 271: 820-821.
14. Doniach D, Roitt IM, 1962 Hakaru Hashimoto. Lancet 1074.
15. Jameson JL, Weetman AP, 2012 Disorders of the Thyroid glands. In: Dan Longo, Anthony Fauci, Dennis Kasper, Stephen Hauser, J. Jameson, Joseph Loscalzo (eds) Harrison’s Principles of Internal Medicine, 18th edition. McGraw Hill Medical Companies. pp. 2911-2939.
16. Amino N, Hagen SR, Yamada N, et al, 1976 Measurement of circulating thyroid microsomal antibodies by the tanned red cell haemagglutination technique: its usefulness in the diagnosis of autoimmune thyroid diseases. Clin Endocrinol (Oxf) 5: 115-125.
17. Brent GA, Davies TF, 2011 Hypothyroidism and thyroiditis. In: Shlomo Melmed, Kenneth S. Polonsky MD, P. Reed MD Larsen, Henry M. Kronenberg MD (eds) Williams Textbook of Endocrinology, 12th Edition, pp 406-439.
18. Amino N, Mori H, Iwatani Y, et al, 1982 High prevalence of transient post-partum thyrotoxicosis and hypothyroidism. N Engl J Med 306: 849-852.
19. Akamizu T, Amino N, DeGroot LJ, 2012 Hashimoto’s thyroiditis. In: Thyroid Disease Manager (http://www.thyroidmanager.org/chapter/hashimotos-thyroiditis/)
20. Hasham A, Tomer Y, 2012 Genetic and epigenetic mechanisms in thyroid autoimmunity. Immunol Res 54: 204-213.
21. Brix TH, Kyvik KO, Hegedüs L. 2000 A population-based study of chronic autoimmune hypothyroidism in Danish twins. J Clin Endocrinol Metab 85:536-539.
22. Menconi F, Monti MC, Greenberg DA, et al, 2008 Molecular amino acid signatures in the MHC class II peptide-binding pocket predispose to autoimmune thyroiditis in humans and in mice. Proc Natl Acad Sci USA 105: 14034-14039.
23. Stefan M, Jacobson EM, Huber AK, et al, 2011 Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon alpha-modulated mechanism. J Biol Chem 286: 31168-31179.
24. Jacobson EM, Huber AK, Akeno N, et al, 2007 A CD40 Kozak sequence polymorphism and susceptibility to antibody-mediated autoimmune conditions: the role of CD40 tissue-specific expression. Genes Immun 8: 205-214.
25. Tomer Y, 2010 Genetic susceptibility to autoimmune thyroid disease: past, present, and future. Thyroid 20: 715-725.
26. Saitoh O, Nagayama Y, 2006 Regulation of Graves’ hyperthyroidism with naturally occurring CD4+CD25+ regulatory T cells in a mouse model. Endocrinology 147: 2417-2422.
27. McLachlan SM, Nagayama Y, Pichurin PN, et al, 2007 The link between Graves’ disease and Hashimoto’s thyroiditis: a role for regulatory T cells. Endocrinology 148: 5724-5733.
28. Burek CL, Talor MV, 2009 Environmental triggers of autoimmune thyroiditis. J Autoimmun 33: 183-189.
29. Duntas LH, 2008 Environmental factors and autoimmune thyroiditis. Nat Clin Pract Endocrinol Metab 4: 454-460.
30. Saranac L, Zivanovic S, Bjelakovic B, et al, 2011 Why is the thyroid so prone to autoimmune disease? Horm Res Paediatr 75: 157-165.
31. Morohoshi K, Takahashi Y, Mori K, 2011 Viral infection and innate pattern recognition receptors in induction of Hashimoto’s thyroiditis. Discov Med 12: 505-511.
32. Giordano C, Stassi G, De Maria R, et al, 1997 Potential involvement of Fas and its ligand in the pathogenesis of Hashimoto’s thyroiditis. Science 275: 960-963.
33. Zaletel K, Gaberšček S, 2011 Hashimoto’s Thyroiditis: From Genes to the Disease. Curr Genomics 12: 576-588.
34. Amino N, Tada H, Hidaka Y, 1999 Postpartum autoimmune thyroid syndrome: a model of aggravation of autoimmune disease. Thyroid 9: 705-713.
35. Katz SM, Vickery AL Jr, 1974 The fibrous variant of Hashimoto’s thyroiditis. Hum Pathol 5: 161-170.
36. Ahmed R, Al-Shaikh S, Akhtar M, 2012 Hashimoto thyroiditis: a century later. Adv Anat Pathol 19: 181-186.
37. Hennessey JV, 2011 Clinical review: Riedel’s thyroiditis: a clinical review. J Clin Endocrinol Metab 96: 3031-3041.
38. Hay ID, 1985 Thyroiditis: a clinical update. Mayo Clin Proc 60: 836–843.
39. Stone JH, Khosroshahi A, Deshpande V, et al, 2012 IgG4-Related disease: recommendations for the nomenclature of this condition and its individual organ system manifestations. Arthritis Rheum 64: 3061-3067.
40. Kakudo K, Li Y, Taniguchi E, et al, 2012 IgG4-related disease of the thyroid glands. Endocr J 59: 273-281.
41. Deshpande V, Huck A, Ooi E, et al, 2012 Fibrosing variant of Hashimoto thyroiditis is an IgG4 related disease. J Clin Pathol 65: 725-728.
42. Tamaki H, Amino N, Kimura M, et al, 1990 Low prevalence of thyrotropin receptor antibody in primary hypothyroidism in Japan. J Clin Endocrinol Metab 71: 1382-1386.
43. Brain L, Jellinek EH, Ball K, 1966 Hashimoto’s disease and encephalopathy. Lancet 2: 512-514.
44. Schiess N, Pardo CA, 2008 Hashimoto’s encephalopathy. Ann N Y Acad Sci 1142: 254-265.
45. Yoneda M, Fujii A, Ito A, et al, 2007 High prevalence of serum autoantibodies against the amino terminal of alpha-enolase in Hashimoto’s encephalopathy. J Neuroimmunol 185: 195-200.
46. Mack LA, Pasieka JL, 2007 An evidence-based approach to the treatment of thyroid lymphoma. World J Surg 31: 978-986.
47. Ahn D, Heo SJ, Park JH, et al, 2011 Clinical relationship between Hashimoto’s thyroiditis and papillary thyroid cancer. Acta Oncol 50: 1228-1234.
Address for correspondence:
Yuji Hiromatsu, Division of Endocrinology and Metabolism,
Department of Medicine, Kurume University School of
Medicine, 67 Asahimachi, Kurume, Fukuoka, 830-0011,
Japan; Tel: +81 942 31 7563; Fax: +81 942 35 8943;
e-mail: yuji@med.kurume-u.ac.jp
Received 03-09-12, Accepted 29-10-12