11st Department of Internal Medicine, Laiko Hospital, Medical School, National and Kapodistrian University of Athens, Greece; 2Section on Endocrinology & Genetics, Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Insitutes of Health (NIH), Bethesda, USA; 3Laboratory of Pathology, National Cancer Institute (NCI), NIH, Bethesda, USA; 4Department of Biological Chemistry, National and Kapodistrian University of Athens, Greece
Medullary thyroid carcinoma (MTC) is a neuroendocrine tumor hereditary in 35% of cases. The most common syndromic form is in the context of the multiple endocrine neoplasia type 2 (MEN 2) syndromes in association with other tumors and due to germline RET mutations. We describe a 57-year-old female patient diagnosed with sporadic MTC. The patient had a history of other neoplasias, such as acute myeloid leukemia, for which she had received chemotherapy, and two other solid tumors, peritoneal mesothelioma and meningioma. Genetic analyses were carried out including whole exome and Sanger sequencing (WES and SS) and loss-of-heterozygosity (LOH) testing for the respective loci. Immunohistochemistry (IHC) was used for the detection of proteins of interest. WES showed two germline variants in the APC and RASAL1 genes confirmed by SS. In MTC tissue only there was a RETvariant identified by SS; germline studies did not show any RETsequence changes. The pattern of tumors in this patient is unusual for either one of the APC- orRASAL1-associated neoplasms and her non-MEN 2-associated MTC contained a RET variant like other sporadic MTCs. As in other patients with more than one genetic variant predisposing to tumors, it is possible that this case represents a unique association.
Acute myeloid leukemia, Medullar thyroid carcinoma, Meningioma, MEN2, Peritoneal mesothelium, RET
INTRODUCTION
Medullary thyroid carcinoma (MTC), a neuroendocrine tumor arising from parafollicular C-cells of the thyroid gland, accounts for 5-10% of all thyroid cancers, with a 1-2% incidence in nodular thyroid disorders.1 MTCs may be either sporadic or hereditary. Hereditary tumors are associated with multiple endocrine neoplasia (MEN) 2 syndrome: MEN2A, MEN2B and familial MTC (FMTC).2 Sporadic forms account for 75% of cases, while MEN2A accounts for a little more than 34% of all hereditary MTCs, with a high (90%)3,4 penetrance.5
MEN2A and MEN2B syndromes have distinctive phenotypes and autosomal dominant inheritance and are due to RET proto-oncogene genetic alterations.3-5 The most common abnormalities that can occur in MEN2A patients are pheochromocytomas (in 40-60% of cases) and hyperparathyroidism (in 10-30% of cases).6,7 MEN2B subjects have no observable parathyroid abnormalities; however, 40-60% of cases develop pheochromocytomas.4 The presence of cutaneous lichen amyloidosis, pruritic plaques located on the upper back, is also reported in some kindreds with MEN2A or FMTC,7 as well as Hirschsprung’s disease in some MEN2A cases.8 FMTC is also an autosomal dominant condition due to genetic alterations in the RET proto-oncogene. It has the least aggressive features9 and is the most common form of hereditary MTC (57.6%).4
We present a patient with multiple endocrine and non-endocrine neoplasias— including MTC—which are not correlated and cannot be included in any of the known clinical syndromes associated with MTC. Genetic analyses revealed the presence of two ‘de novo’ germline mutations, one in the adenomatous polyposis coli (APC) gene and the other in the RAS Protein Activator Like 1 (RASAL1) gene as well as one somatic mutation in the RET gene, which however is not yet known to be related to the MTC.
CASE PRESENTATION
A 57-year-old female was followed for bilateral MTC diagnosed 4 years ago. At that time she was submitted to total thyroidectomy as well as lymph nodes excision (7/17 positive) of the left cervical compartments. Histology showed that the carcinoma was extended with diffuse infiltration of both thyroid lobes and the paratracheal fat tissue (stage, T3mN1M0). Imaging had showed no distal metastases. Pre-operative serum calcitonin levels were high at 140 μU/ml baseline and 244 after stimulation with calcium, while levels post-operatively were 14 μU/ml with normal serum CEA levels.
She presented to the outpatient endocrinology clinic of our hospital for her follow-up. Her family medical history was non-contributory. Her personal medical history included acute myeloid leuchemia (AML) diagnosed at the age of 41 years old and treated with chemotherapy (she had received initially two cycles of idarubicine/cytarabine with complete regression—however, she developed Sweet syndrome—and two cycles of floudarabin and cytarabine); since September 2002 she is considered in remission. Her medical history was also marked by a surgical exeresis of a peritoneal mesothelium at the age of 43 years old and a meningioma of 2×1.6×1.7 cm diagnosed at the age of 53 years old for which she has constant follow-up. Τhe patient was also known to suffer from polyglandular autoimmune syndrome type 3 (PASIIIc) including Hashimoto’s thyroiditis, autoimmune gastritis, alopecia universalis and relapsing perichondritis of the ear, while recently rheumatoid arthritis was also diagnosed.
METHODS
Genetic screening for RET gene variant
A peripheral blood sample was drawn in EDTA for molecular investigations after informed consent was obtained from the patient. DNA was extracted from peripheral blood leucocytes according to commercially available protocols (QIAGEN, Valencia, CA, USA). Genomic DNA fragments encompassing regions in the following exons: 5, 8, 10, 11, 13, 14, 15, previously found to contain activating missense RET mutations related to MTC, were amplified by polymerase chain reaction (PCR) and analyzed by classical bidirectional Sanger sequencing using BigDye Terminator V3.0 (Applied Biosystems).
Given this result, DNA was then extracted from paraffin-embedded thyroid tissue and DNA fragments encompassing all exons of the RET gene were amplified by PCR, while direct sequencing of the purified fragment was performed using BigDye Terminator V3.0 (Applied Biosystems).
RESULTS
No mutation in the aforementioned exons of the RET gene was detected in the blood sample. Interestingly, in the MTC tissue the variant c.1546C>T (ENST00000355710.7, NM_020975) was detected (Figure 1). The c.1546C>T leads to the replacement of the non-polar hydrophobic amino acid proline by the polar hydrophilic serine (p.P516S). This mutation is not reported either in gnomAD or in 1000GP. Further genetic analyses of blood samples, including SNP microarrays (comparative genomic hybridization; CGH) and WES (whole exome sequencing), were also performed as part of our diagnostic work-up.
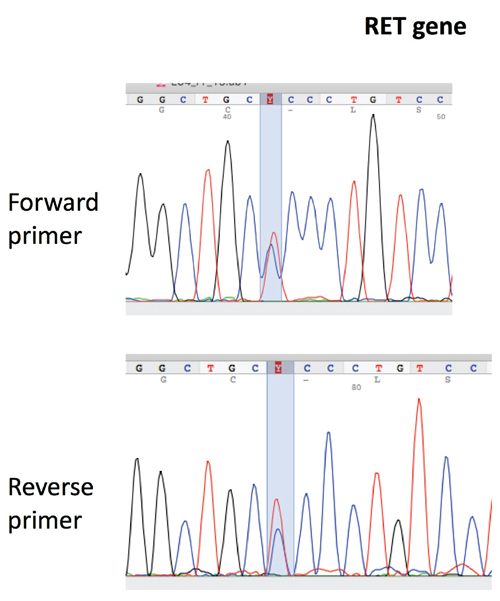
Figure 1. Electropherograms showing the analysis of the RET gene (Reference Sequence: NM_001193520) in the medullar thyroid carcinoma tissue showing the substitution of cytosine to thymine (c.1546C>T).
Genetic analyses with CGH and WES
CGH showed no abnormalities. However, WES revealed the following two ‘novel’ mutations. One is heterozygous (c.3307 A>T) in exon 16 of the APC gene (NM_000038.5), leading to the replacement of arginine (R), a positively charged hydrophilic amino acid, by the hydrophobic tryptophan (W) (p.R1103W). The second (c.1613G>A) is also a heterozygous mutation in exon 16 of the RASAL1 gene (NM_001193520). The c.1613 G>A RASAL1 mutation, which leads to the amino acid replacement of arginine (R) by histidine (H) (p.R538H), was already described in the gnomAD database, with a frequency of 0.0037%.
In silico analysis
Two sites of prediction of the functional effects of gene polymorphism were used: Polyphen (http://genetics.bwh.harvard.edu/pph2/), and SIFT (http://sift.jcvi.org/).
In silico analysis of the c.3307 A>T APC mutation predicts that this variant is probably damaging to the protein structure/function. The in silico characterization of the c.1613G>A RASAL1 mutation is inconsistence in its predictions as to whether or not the variant is damaging to the protein function/structure.
Immunochemistry
Paraffin-embedded tissues from MTC were stained for APC and RASAL1 protein expression (Figures 2, 3). MTC tissue showed an intense and diffuse staining for APC protein, whereas staining for RASAL1 was faintly positive.
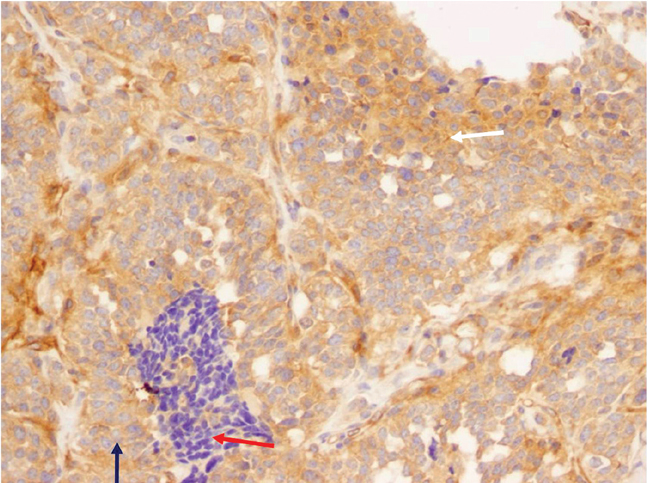
Figure 2. IHC analysis of paraffin-embedded MTC tissue revealed diffuse positive staining (20×). White arrow: Tumor cells with intense cytoplasmic immunoreactivity. Red arrow: Lymphocytic infiltration which is clearly negative (negative control). Blue arrow: Normal pattern for normal follicular cells: mostly negative—the nucleus is negative and the cytoplasm very weakly positive.

Figure 3. Faint staining of the RASAL1 protein in the IHC analysis in the medullar thyroid carcinoma tissue (20×). Normally the RASAL1 gene is very highly expressed in thyroid cells since the thyroid along with the brain and adrenal glands are the sites of highest expression. White arrow: Cluster of cells that are positive but that do not look like tumor cells. Red arrow: Lymph nodes: a negative control.
DISCUSSION
Herein, we report two novel germline mutations detected in the APC and RASAL1 genes, identified through WES, in a patient presenting with multiple neoplasias such as MTC, AML, peritoneal mesothelioma and meningioma; we also found a somatic missense RET mutation in the MTC tissue.
Neither the germline heterozygous variants in the APC gene nor the RASAL1 gene have been reported previously as pathogenic variants. Of note, the APC mutation has not been described as either pathogenic or benign in people of European or Africo-American origin participating in the NHLBI exome sequencing project, while the RASAL1 mutation was already described in the gnomAD database, with a frequency of 0.0037%.
The APC gene is an oncosuppressive gene involved in the WNT signaling pathway. Approximately 25% of pathogenic variants in the APC gene occur de novo.10 The p.R1103W variant is a non-conservative amino acid substitution and occurs in a position within the 15-amino acid, repeat β-catenin binding domain that is conserved across species. Pathogenic truncating variants in the APC gene are most commonly associated with autosomal dominant familial adenomatous polyposis (FAP) characterized by early-onset presentation of hundreds of adenomatous colon and rectal polyps as well as duodenal, stomach, thyroid, pancreatic, brain and liver cancers.10,11 Variable presentations of FAP include also Gardner syndrome (osteomas and soft tissue tumor) and Turcot syndrome (colonic polyposis and central nervous system tumors like medulloblastoma and malignant glioma).12
Rare somatic variants have been reported in individuals with hepatoblastoma and gastric cancer (MIM 611731). Interestingly, APC staining was found strongly positive in the MTC tissue with characteristic cytoplasmic localization; it could be hypothesized that the presence of this mutation could abolish the entrance of APC into the nucleus, keeping it mainly in the cytoplasm. The RASAL1 gene encodes the RAS GTP-ase which serves as an inhibitory regulator of the Ras-cyclic AMK pathway that is expressed in a limited number of tissues, with the highest expression in the adult brain, thyroid gland and adrenal gland (MIM 604118).13
Follicular variant papillary thyroid cancer presents a higher frequency among individuals with RASAL1 variants compared to those without.13 Therefore, germline RASAL1 variants may be relatively frequent in patients with apparently sporadic thyroid carcinoma with follicular features.13 The R538H variant is a conservative amino acid substitution which is not likely to impact secondary protein structures as these residues share similar properties. The substitution occurs at a position that it is not conserved among species. In silico analysis for this variant was not clear. Thus this variant was interpreted as being of uncertain significance and could be related to the thyroid cancer reported in this patient. RASAL1 staining in the tissue was less intense compared to APC.
The somatic variant in the RET detected in the MTC tissue has not been previously described. The co-existence of multiple neoplasias without common histological and embryological origin in this patient cannot be included in a specific genetic pattern or clinical syndrome according to the existing data in the literature. A secondary cause cannot be excluded in this patient already diagnosed with AML and treated with chemotherapy as it is known that prior exposure to chemotherapy can result in various types of somatic mutation or even germ mutation, thus predisposing to a second neoplasm.14-16 Interestingly, there are some rare cases reporting occurrence of papillary thyroid cancer and meningioma in patients treated for acute lymphocytic leukemia or some years after bone marrow transplantation, especially during childhood.17,18
CONCLUSION
Patients with ‘mixed or atypical’ phenotypes often present a diagnostic challenge. Genetic counseling in these cases is difficult because there is no specific genetic pattern. Here we report the case of a patient with multiple neoplasias including MTC which are not correlated and cannot be included in any known clinical syndrome. Genetic analyses showed that the patient presents two new germline mutations ‘de novo’, one in the adenomatous polyposis coli (APC) gene and the other in the RAS Protein Activator Like 1 gene as well as one somatic mutation in the RET gene, which however have never before been identified in MTC. The mutations in the APC and RASAL1 genes are probably involved in thyroid cancer development, along with the somatic mutation in the RET gene. In addition, the mesothelioma and meningioma are likely related to mutations in the APC gene. Both germline mutations were characterized as variants of uncertain significance and they have not been previously described as pathogenic.
DECLARATION OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
FUNDING SOURCES
This research was supported in part by the Intramural Research Program NICHD, NIH.
PATIENT CONSENT
Written informed consent was obtained from the patient.
ACKNOWLEDGEMENTS
None.
REFERENCES
1. Ball DW, 2000 Medullary thyroid carcinoma. Thyroid cancer. Springer: 365-381.
2. Marquard J, Eng C 2015 Multiple endocrine neoplasia type 2. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N, (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017.
3. Moley JF, 2003 Medullary thyroid carcinoma. Curr Treatm Options Oncol 4: 339-347.
4. Romei C, Mariotti S, Fugazzola L, et al, 2010 Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol 163: 301-308.
5. Wohllk N, Schweizer H, Erlic Z, et al, 2010 Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab 24: 371-387.
6. DeLellis R, Wolfe H, Gagel R, et al, 1976 Adrenal medullary hyperplasia. A morphometric analysis in patients with familial medullary thyroid carcinoma. Am J Pathol 83: 177.
7. Gagel RF, Tashjian AH, Jr, Cummings T, et al, 1988 The clinical outcome of prospective screening for multiple endocrine neoplasia type 2a. N Engl J Med 318: 478-484.
8. Verdy M, Weber AM, Roy CC, Morin CL, Cadotte M, Brochu P, 1982 Hirschsprung’s disease in a family with multiple endocrine neoplasia type 2. J Pediatr Gastroenterol Nutr 1: 603-608.
9. Nose V, 2011 Familial thyroid cancer: a review. Modern Pathol 24:19–33.
10. Jasperson KW, Tuohy TM, Neklason DW, Burt RW, 2010 Hereditary and familial colon cancer. Gastroenterology 138: 2044-2058.
11. Jasperson K, Burt RW, 2015 The Genetics of Colorectal Cancer. Surg Oncol Clin N Am 24: 683-703.
12. Jasperson KW, 2012 Genetic testing by cancer site: colon (polyposis syndromes). Cancer J 18: 328-333.
13. Ngeow J, Ni Y, Tohme R, Song Chen F, Bebek G, Eng C, 2014 Germline alterations in RASAL1 in Cowden syndrome patients presenting with follicular thyroid cancer and in individuals with apparently sporadic epithelial thyroid cancer. J Clin Endocrinol Metab 99: 1316-1321.
14. Kubota M, Lin YW, Hamahata K, et al, 2000 Cancer chemotherapy and somatic cell mutation. Mutat Res 470: 93-102.
15. Swisher EM, Harrell MI, Norquist BM, et al, 2016 Somatic Mosaic Mutations in PPM1D and TP53 in the Blood of Women With Ovarian Carcinoma. JAMA Oncol 2: 370-372.
16. Glen CD, Dubrova YE, 2012 Exposure to anticancer drugs can result in transgenerational genomic instability in mice. Proc Natl Acad Sci U S A 109: 2984-2988.
17. Barg E, Godziński J, Wikiera B, Głab E, Jeleń M, 2009 Papillary thyroid carcinoma and meningioma in 17-year-old boy - a late effect of acute lymphoblastic leukemia treatment or a new disease? Case report and literature review. Pediatr Endocrinol Diabetes Metab 15: 51-54.
18. Gow KW, Lensing S, Hill DA, et al, 2003 Thyroid carcinoma presenting in childhood or after treatment of childhood malignancies: An institutional experience and review of the literature. J Pediatr Surg 38: 1574-1580.
Address for correspondence:
Eva Kassi; Medical School, National and Kapodistrian University of Athens, 75 Mikras Asias Str., Goudi, 11527 Athens, Greece; Tel.: +302107462699, +306974856280, Fax: +302107462703, E-mail: ekassi@med.uoa.gr
Received: 11-10-2017, Accepted: 17-10-2017