Αnti-Müllerian Ηormone gene
1Denizli State Hospital, Pediatric Endocrinology Clinic, Denizli, 2Denizli State Hospital, Pediatric Surgery Clinic, Denizli, 3Ege University Faculty of Medicine, Medical Genetics Department, İzmir, Turkey
BACKGROUND
AND OBJECTIVE: Persistent Müllerian duct syndrome (PMDS) is a relatively rare
form of 46,XY disorder of sex development caused by the failure of formation,
release or action of anti-Müllerian hormone (AMH) in intrauterine life. In this
report we describe a case diagnosed with PMDS with a novel homozygous mutation
in the AMH gene.
CASE REPORT: A 4-month-old male presented with
bilateral cryptorchidism and normal external genitalia. The laboratory
examination revealed normal gonadotropin levels for his age (FSH: 0.91 mIU/mL,
LH: 1.23 mIU/mL, testosteron <0.13 ng/mL, respectively). AMH was
undetectable (<0.01 ng/mL). Ultrasonography (USG) revealed absence of the
left gonad and an intraabdominally located right gonad. Laparoscopy
demonstrated the presence of a rudimentary uterus and fallopian tubes.
Karyotyping revealed a normal 46,XY karyotype. Molecular genetic analysis
demonstrated a novel homozygous mutation [p.C526F (c.1577G>T)] in the AMH
gene.
CONCLUSION: PMDS should be kept in mind in all cases with bilateral crytorchidism.
Orchidopexy and resection of Mulletian duct derivates, exercising extra caution
with regard to maintaining vascular supply to the testis, is the recommended
approach
AMH, Crytorchidism, Disorders of sex development
INTRODUCTION
Anti-Müllerian hormone (AMH), which is a member of the transforming growth factor-β superfamily, is secreted mainly by immature Sertoli cells at the time of sex differentiation in fetal life (i.e., at the end of the seventh gestation week).1 The main role of fetal AMH is the regression of Müllerian ducts (fallopian tubes, uterus, and upper vagina).1,2
Persistent Müllerian duct syndrome (PMDS) is a rare form of a 46,XY disorder of sex development, in which remnants of Müllerian ducts are seen in phenotypically normal males.2 The syndrome is caused either by a mutation in the AMH gene or the AMH receptor gene (AMHR-II), which are located on chromosomes 19p13.3 and 12q13.13, respectively. AMH gene mutations are referred to as PMDS type I and AMHR-II mutations are referred to as PMDS type II.3 Mutations within these two genes have been detected in 85% of patients with PMDS, with the causative genes unidentified in the remaining 15% of cases. Although autosomal recessive, autosomal dominant, and X-linked inheritance patterns have been reported in PMDS, most of the reported cases were isolated.2-4
The AMH gene was mapped by Cohen-Hauguenaure in 1987.5 Since then, more than 200 PMDS cases have been reported in the literature.1-4,6
In this case report, we describe the clinical features of a case of PMDS type I with a novel AMH mutation.
MATERIAL AND METHODS
Case
The study was approved by the local ethics committee of Denizli State Hospital, Turkey. Written informed consent for the study was given by the parents of the patient.
A 4-month-old boy was admitted to the pediatric surgery clinic with the diagnosis of bilateral nonpalpable testes. There was second-degree consanguinity between his parents. A physical examination revealed a normal prepubertal sized penis (3.5 cm) and a normally developed scrotum. However, the gonads were bilaterally nonpalpable. The results of other systematic examinations were normal.
The serum levels of gonadotropins and testosterone were normal for the patient’s age (Table 1). The serum AMH level was undetectable (<0.01 ng/mL). A human chorionic gonadotropin (hCG) test (1500 IU/m2/dose, 3 consecutive days) was performed. Subsequently, the serum testosterone level increased to 7.96 ng/mL. A karyotype analysis of peripheral blood revealed a 46,XY karyotype. An ultrasonographic evaluation revealed no left gonad and an intraabdominally located right gonad. The findings of abdominal magnetic resonance imaging were compatible with those of the ultrasonography.
Laparoscopy revealed the presence of a rudimentary uterus at the midline and symmetrical fallopian tubes. The morphology of the gonadal structures was consistent with the testes being in a pseudo-ovarian position (i.e., the structures were attached to the fimbrial end of the fallopian tubes) (Figure 1). An exploratory laparotomy/excision was planned for the patient. Staged bilateral orchidopexy (Fowler-Stephens technique) was performed and the Müllerian structures were removed.
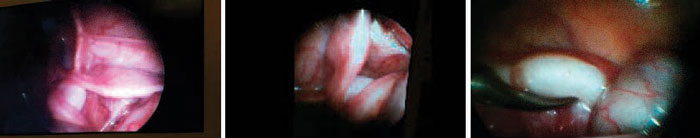
Figure 1. Laparascopic view of the patients.
A novel homozygous p.C526F (c.1577G>T)
mutation on an evolutionary conserved amino acid was detected in the patient
(Figure 2). According to PolyPhen (score: 1), SIFT (score: 0), and
MutationTaster (p-value: 1) in silico analysis software programs, this novel
mutation was pathogenic. The mutation was located on the TGF-beta domain of the
protein. This domain is important for interactions with other proteins.
Mutations on adjacent amino acids (525th and 527th) have been reported
previously, all of which were disease-causing mutations.7

Figure 2. The p.C526F (c.1577G>T) mutation detected in the patient.
Endocrine evaluation
Serum levels of FSH, LH, testosterone, and cortisol were determined using a chemiluminescent immunoassay (Architect; Abbott Diagnostics). AMH was measured by an electrochemiluminescence immunoassay (ECLIA) kit (Cobas 6000 e601; Roche Diagnostics). 17-hydroxyprogesterone was measured by RIA (PC-RIA.MAS Stratec, Germany).
Molecular analysis
DNA from the patient was isolated from peripheral blood leucocytes. A mutation analysis was then performed in the medical genetics department of Ege University, İzmir, Turkey. The AMH gene mutation analysis was performed by sequencing the coding exons of the gene. Genomic DNA was isolated from peripheral blood cells using standard techniques and PCR was performed. All PCR products were sequenced by the dye termination method using a DNA sequencing kit (Perkin-Elmer, CA, USA) and analyzed using an ABI Prism 3100 sequence analyzer (Applied Biosystems, CA, USA).
DISCUSSION
In a male human fetus, testosterone, dihydrotestosterone, and AMH are required for normal sex differentiation. AMH is produced by Sertoli cells by the end of the seventh gestational week and causes regression of the Müllerian ducts. Testosterone promotes the differentiation of the Wolffian ducts into the epididymis, vas deferens, and seminal vesicles. Dihydrotestosterone induces differentiation of external genitalia.1
Due to a deficiency of AMH or the action of AMH, PMDS patients have both Wolffian and Müllerian ductal structures but normally developed external genitalia and Wolffian structures.1,2 In PMDS patients, remnants of the Müllerian duct (i.e., fallopian tubes, uterus, and the upper part of the vagina) are present in genotypically and phenotypically normal males. The main clinical characteristics are cryptorchidism and transverse testicular ectopia.1,2
PMDS can be classified into three anatomical types, depending on the location of the testes and Müllerian ductal structures. The most common type is the “male form”, which is found in 80-90% of cases. In this type, at least one testis is present in the scrotum. In addition, the uterus and ipsilateral tubes are either present in the sac or can be drawn. The second (least common) type is the “female form”, seen in 10-20% of cases. This type of PMDS is characterized by the presence of testes in the pseudo-ovarian position. The testes are attached to the fimbrial end of the fallopian tubes and the uterus is in the normal female pelvic position. In the third type, known as “transverse testicular ectopia”, both testes are present on the same side, either in the inguinal canal or scrotum.8 The clinical features in the present case were compatible with the second (female form) type of PMDS.
AMH gene mutations (PMDS type I) are responsible for approximately 45-52% of genetically proven PMDS cases. AMHR2 gene mutations (PMDS type II) are responsible for the remaining 40% of cases.3,4 Patients with AMH or AMHR2 gene mutations show no differences in the anatomical phenotype. However, patients with AMH gene defects have low or undetectable AMH levels, whereas those with AMHR mutations have normal or elevated levels of AMH. Laparoscopy is the preferred diagnostic procedure in cases of a suspicion of AMHR2 mutations. The AMH gene is localized on the 19p13.3 chromosome and consists of 5 exons. Most AMH mutations are homozygous and located on exon 1 and exon 5.1-4 To the best of our knowledge, as of June 2016 approximately 57 AMH mutations have been reported.1,6 The homozygous p.C526F (c.1577G>T) mutation detected in the present case is a novel mutation.
The risk of malignancy in an ectopic testis in a case of PMDS is similar to that in a cryptorchid testis in a healthy male. Germ cell tumors (embryonal carcinoma, seminomas, yolk sac tumors, and teratomas) are the most common types of tumors reported in the testes.8,9 Tumors of Müllerian ductal derivatives have also been reported.10
The treatment of PMDS is surgical. There are some controversies regarding the management of PMDS patients. Bringing the testes down to a normal position is usually difficult in PMDS cases due to the shortness of the spermatic cord and the location of the vasa deferentia in the mesosalpinx or uterine wall. During surgery, the residual Müllerian ductal structures should be preserved to protect the vascular supply of the testis and vas deferens. However, malignant tumors of the Müllerian ducts have been reported.8,9,11 Thus, some authors have proposed the adoption of a more aggressive surgical approach, which involves removing the Müllerian ductal structures.8,9,11
To conclude, in all cases of bilateral cryptorchidism, the possibility of PMDS should be borne in mind. Genetic counselling should be considered in cases of parental consanguinity.
GRANT OR FELLOWSHIP SUPPORT
None
CONFLICT OF INTEREST
None
REFERENCES
1. Josso N, Rey RA, Picard JY, 2013 Anti-Müllerian hormone: a valuable addition to the toolbox of the pediatric endocrinologist. Int J Endocrinol 2013: 674105.
2. Elias-Assad G, Elias M, Kanety H, Pressman A, Tenenbaum-Rakover Y, 2016 Persistent Müllerian duct syndrome caused by a novel mutation of an anti-Müllerian hormone receptor gene: Case presentation and literature review. Pediatr Endocrinol Rev 13: 731-740.
3. Belville C, Van Vlijmen H, Ehrenfels C, et al, 2004 Mutations of the anti-Müllerian hormone gene in patients with persistent Müllerian duct syndrome: biosynthesis, secretion, and processing of the abnormal proteins and analysis using a three-dimensional model. Mol Endocrinol 18: 708-721.
4. Josso N, Belville C, di Clemente N, Picard JY, 2005 AMH and AMH receptor defects in persistent Müllerian duct syndrome. Hum Reprod Update 11: 351-356.
5. Cohen-Haguenauer O, Picard JY, Mattei MG, et al, 1987 Mapping of the gene for anti-Müllerian hormone to the short arm of human chromosome 19. Cytogenet Cell Genet 44: 2-6.
6. Nishi MY, Domenice S, Maciel-Guerra AT, et al, 2012 Analysis of anti-Müllerian hormone (AMH) and its receptor (AMHR2) genes in patients with persistent Müllerian duct syndrome. Arq Bras Endocrinol Metabol 56: 473-478.
7. Imbeaud S, Belville C, Messika-Zeitoun L, et al 1996 A 27 base-pair deletion of the anti-Müllerian type II receptor gene is the most common cause of the persistent müllerian duct syndrome. Hum Mol Genet 5: 1269-1277.
8. Saleem M, Ather U, Mirza B, et al, 2016 Persistent Müllerian duct syndrome: A 24-year experience. J Pediatr Surg 51: 1721-1724.
9. Ju X, Li Z, Zhang C, et al, 2013 Clinical aspects and molecular genetics of persistent Müllerian duct syndrome associated with transverse testicular ectopia: report of three cases. Urol Int 90: 83-86.
10. Farikullah J, Ehtisham S, Nappo S, Patel L, Hennayake S, 2012 Persistent Müllerian duct syndrome: lessons learned from managing a series of eight patients over a 10-year period and review of literature regarding malignant risk from the Müllerian remnants. BJU Int 110: E1084-1089.
11. Shalaby MM, Kurkar A, Zarzour MA, Faddan AA, Khalil M, Abdelhafez MF, 2014 The management of the persistent Müllerian duct syndrome. Arab J Urol 12: 239-244.
Address for correspondence:
Ayca Altincik, MD, Denizli State Hospital, Pediatric Endocrinology Clinic, 20100, Denizli, Turkey, Tel.: +90 5337498817, E-mail: ayca.altincik@deu.edu.tr
Received:20-03-2017, Accepted:28-04-2017