1Division of Endocrinology, Metabolism and Diabetes, First Department of Pediatrics, National and Kapodistrian University of Athens Medical School, ‘Aghia Sophia’ Children’s Hospital, Athens, Greece; 2Division of Endocrinology and Metabolism, Center of Clinical, Experimental Surgery and Translational Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece
Glucocorticoids play a fundamental role in many physiologic functions and contribute substantially to the achievement of homeostasis. These pleiotropic glucocorticoid actions are mediated by a ubiquitously expressed transcription factor, the human glucocorticoid receptor (hGR), which may influence the transcription rate of numerous target genes, interact with other transcription factors, trigger the activation of several kinase pathways or modulate mitochondrial DNA expression. Any genetic defects in the NR3C1gene that encodes the hGR may cause Primary Generalized Glucocorticoid Resistance or Hypersensitivity Syndromes, two rare allostatic endocrinologic conditions characterized by partial impaired tissue sensitivity to glucocorticoids. However, there are patients who present with clinical manifestations suggestive of the above syndromes and do not harbor an inactivating or activating point mutation, insertion or deletion in the NR3C1gene. In these cases, several other factors might influence the glucocorticoid signal transduction. In this review, we discuss the numerous glucocorticoid functions and the multiple hGR isoforms, we present the genomic, nongenomic and mitochondrial glucocorticoid signaling cascade and we summarize the clinical manifestations and pathogenesis of Primary Generalized Glucocorticoid Resistance or Hypersensitivity Syndromes. Finally, we speculate that the next generation sequencing technologies will undoubtedly enable us to gain a deeper understanding of the GR “interactome”.
Glucocorticoid receptor, Glucocorticoid signaling, Glucocorticoid hypersensitivity, Glucocorticoid resistance, Glucocorticoids, NR3C1 mutations
GLUCOCORTICOIDS: STEROID
HORMONES WITH PLEIOTROPIC FUNCTIONS
Natural glucocorticoids (cortisol in humans and corticosterone in rodents) are steroid hormones essential for life and play a crucial role in the achievement of resting and stress-related homeostasis.1-4 These lipophilic molecules are derived from cholesterol through serial enzymatic reactions that lead to the production of all steroid hormones.5 For many years it was believed that glucocorticoids were exclusively synthesized in endocrine cells located in the zona fasciculata of the adrenal cortex.4,5 Recent evidence, however, points to the presence of extra-adrenal sites of glucocorticoid production, such as the intestine, thymus and skin.6 Following biosynthesis, glucocorticoids are released into the systemic circulation in an ultradian, circadian and stress-related fashion.7,8 The ultradian secretion of glucocorticoids occurs within a short time frame and is not influenced by neuronal input from the hypothalamic suprachiasmatic nucleus (SCN).9 The circadian pattern of glucocorticoid secretion is characterized by high circulating concentrations of these hormones early in the morning and low concentrations in the evening.7 Indeed, light-activated SCN neurons send projections to the paraventricular nucleus (PVN) neurons of the hypothalamus, which synthesize and release corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) into the hypophysial portal system.10 CRH and AVP then reach the anterior lobe of the pituitary gland and trigger synergistically the production and secretion of adrenocorticotropin hormone (ACTH).1-5 Finally, ACTH activates the biosynthetic pathway of glucocorticoids, while it also controls androgen and aldosterone secretion by the zona reticularis and zona glomerulosa, respectively, in the adrenal cortex.5 In addition to the SCN-activated hypothalamic-pituitary-adrenal (HPA) axis, the circadian pattern of glucocorticoid release is influenced by the SCN-derived autonomic innervation of the adrenal glands as well as by circadian clocks located in the adrenal cortex.11 Under acute stress, glucocorticoids are secreted in high amounts and cause a transient desynchronization of the central and peripheral clocks, which is effectively restored within a few days.8 However, under chronic stress this resynchronization may not occur, leading to several metabolic, inflammatory, mood and malignant disorders.8
Glucocorticoids substantially influence myriad physiologic functions.1-4,7,8 They maintain tight control over cardiovascular tone, display catabolic actions in the liver, muscle and adipose tissue and exert potent anti-inflammatory and immunosuppressive effects.1-4,7,8 Moreover, glucocorticoids play a fundamental role in growth, development, reproduction, behavior and cognition. They are also involved in a very large number of cellular processes, such as proliferation, differentiation and programmed cell death (apoptosis).1-4,7,8 Interestingly, a growing body of evidence suggests that glucocorticoids can cause epigenetic alterations, such as changes in the methylation status of several cytosine-guanine dinucleotides (CpG) within a number of promoter regions, by changing the expression and/or activity of enzymes involved in the methylation/demethylation process.12 All these pleiotropic effects of glucocorticoids are mediated by the glucocorticoid receptor (GR), which functions as a ligand-activated transcription factor influencing the transcription rate of approximately 20% of the human genome.13,14 Based on the above-discussed immunomodulating effects, synthetic glucocorticoid analogues are worldwide the cornerstone in the treatment of numerous inflammatory, autoimmune and malignant diseases.15
GLUCOCORTICOID RECEPTOR: FROM THE NR3C1 GENE TO PROTEIN ISOFORMS
The human GR (hGR) is a member of the steroid hormone receptor family of the nuclear receptor superfamily of transcription factors.13 It is encoded by the NR3C1 gene (chromosome 5) and consists of 10 exons. Exon 1 contains multiple tissue-specific non-coding DNA regions, whereas exons 2-9α/9β form the protein expression region.13,16-18 Through alternative splicing, insertions or deletions and via alternative translation initiation, the NR3C1 gene finally encodes multiple protein isoforms with distinct properties in terms of localization and activity13,16-18 (Figure 1).
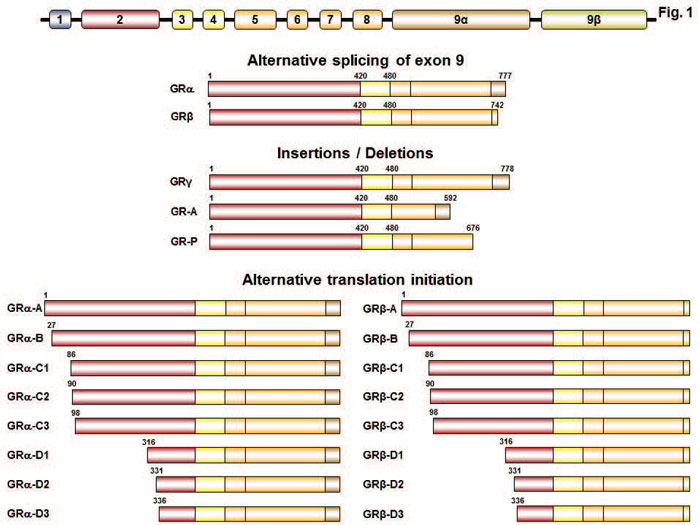
Figure 1. Genomic DNA, protein isoforms and functional domains of hGR. The NR3C1 gene consists of 10 exons. Alternative splicing of exon 9 generates the two main protein isoforms, hGRα and hGRβ. hGRγ, hGR-A and hGR-P are generated by insertions or deletions in the NR3C1 gene. The initiation of hGRα mRNA translation might occur from 8 alternative sites giving rise to 8 different protein isoforms. We hypothesize that the same mechanism occurs in the case of hGRβ. GR: glucocorticoid receptor.
The alternative splicing of exon 9 generates the two main receptor isoforms, termed hGRα and hGRβ.13,16-18 These isoforms share the first 727 amino acids but display structural differences beyond this amino acid position. hGRα contains an additional 50 amino acids, whereas hGRβ is composed of 15 additional non-homologous amino acids. This latter characteristic confers distinct properties on hGRβ.13,16-18 Indeed, hGRβ does not bind natural or synthetic glucocorticoids; it resides in the nucleus of certain cell types, such as epithelial cells and neutrophils and exerts inhibitory effects on the transcriptional activity of the hGRα through well-studied molecular mechanisms.19-21 However, given that studies from several groups have demonstrated that hGRβ can directly induce or repress a number of target genes,22-25 while Chatzopoulou and collaborators recently revealed that GRβ does not have transcriptional activity in zebrafish,26 it is evident that the specific functional role of GRβ remains to be elucidated. New evidence suggests that hGRβ has an important role in insulin signaling and might be involved in gluconeogenesis and inflammation in mouse liver.27,28 Furthermore, this isoform was found to participate in the molecular pathways of glioma formation as well as in the migration of bladder cancer cells.29-31 In addition to in vitro and animal experiments, studies in humans have shown that hGRβ expression levels are associated with glucocorticoid resistance in severa l inflammatory diseases (ulcerative colitis, rheumatoid arthritis, systemic lupus erythematosus, asthma), hematologic malignancies (acute lymphoblastic leukemia, chronic lymphocytic leukemia) as well as in many mood disorders (major depression, schizophrenia).18,32 In contradistinction to hGRβ, the hGRα isoform is ubiquitously expressed in every tissue except the SCN of the hypothalamus, is primarily localized in the cytoplasm of target cells, binds natural and synthetic glucocorticoids and influences the transcription of a large number of glucocorticoid-responsive genes in a positive or negative fashion.13,16-18There are three additional splice GR isoforms, termed GR-γ, GR-A and GR-P, which were found to reside in cancer cells and later in normal tissues.18 GR-γ is formed by an insertion of an arginine residue at amino acid position 452 in the ligand-binding domain (LBD) of the receptor.33 This isoform is expressed in several tissues, binds glucocorticoids with similar affinity to that of the wild-type receptor, but displays 50% transcriptional activity compared to hGRα.34 GR-γ expression has been associated with glucocorticoid resistance in acute lymphoblastic leukemia, small cell lung carcinoma and pituitary corticotroph adenomas.34,35 GR-A lacks a large fragment lying between amino acid positions 490 and 674 (exons 5-7). This splice variant is unable to bind glucocorticoids because of a defective LBD.18 Finally, GR-P lacks exons 7 and 8 and does not bind glucocorticoid hormones; it is expressed in normal tissues and is over-expressed in non-Hodgkin lymphoma, acute lymphoblastic leukemia and multiple myeloma.18,36 This receptor isoform influences hGRα-induced transcriptional activity in a cell-type specific fashion37,38 (Figure 1).
More than a decade ago, Lu and Cidlowski proposed the presence of an additional cohort of GR isoforms generated by alternative initiation of GRα mRNA translation.39 They showed that eight AUG start codons located in exon 2 of the GR transcript might give rise to eight receptor isoforms, termed GRα-A, -B, -C1, -C2, -C3, -D1, -D2 and -D3, with progressively shorter N-terminal domains (NTDs).39 We speculate that the same mechanism might occur in GRβ, GR-γ, GR-A and GR-P. The eight GRα isoforms bind to glucocorticoids with similar affinity. Following ligand-induced activation, they also have a similar capability to bind to the promoter regions of target genes.18 However, these GRα translational variants have distinct transcription properties. Each isoform, when expressed in Jurkat T-lymphoblastic leukemia cells or U2OS osteosarcoma cells, was shown to influence the transcription of a specific set of genes.40,41 Only a small percentage of target genes (less than 10%) was found to be commonly regulated by these isoforms.40,41 Not only do they have a unique transcription profile, but they also possess different subcellular localization. For example, GRα-D was found constitutively in the nuclear compartment, whereas GRα-A, GRα-B and GRα-C were seen to reside primarily in the cytoplasm and to translocate to the nucleus following ligand-induced activation.39 The GRα translational isoforms also differ in terms of tissue expression.18 GRα-B was found mostly in the thymus and colon, whereas GRα-C was highly expressed in the lung, pancreas and colon.39 GRα-D was more abundant in the spleen and bladder.39 The molecular mechanisms underlying tissue expression still remain under investigation.
GENOMIC, NONGENOMIC AND MITOCHONDRIAL GR SIGNALING
At the cellular level, glucocorticoids exert their genomic, nongenomic and mitochondrial effects through activation of their cognate receptor.2,4,13,18 In the absence of glucocorticoids, GR resides primarily in the cytoplasm forming a multiprotein complex that also contains heat shock proteins (HSP90, HSP70) and immunophillins (FKBP51 and FKBP52).42 This complex enables GR to have a ligand-friendly conformation and masks important amino acid sequences of the receptor, nuclear localization signal 1 and 2 (NLS1 and 2), thereby preventing the nuclear translocation of the ligand-unbound receptor.43 Upon ligand binding, GR undergoes an appropriate conformational change that helps it to dissociate from the multiprotein complex and then translocate to the nucleus. Within the nucleus, GR binds, as a homo- or heterodimer, to specific DNA sequences located in the promoter regions of glucocorticoid-responsive genes.2,4,13,18 These sequences, termed glucocorticoid response elements (GREs), consist of two 6-nucleotide half-sites separated by three nucleotides (GGAACAnnnTGTTCT).44 Following GR binding to GREs, the resultant conformational change of the GR dimer leads to the recruitment of co-activators and chromatin-remodeling complexes, which induce the activity of RNA polymerase II, thereby enabling gene transcription.18 Important findings have shed light on the molecular mechanisms underlying GR-mediated gene repression.45 Surjit and collaborators proposed that GR dimers might bind to negative GREs (nGREs) and recruit corepressors, such as NCoR1 and SMRT, as well as histone deacetylases, leading to repression of several target genes.45 nGRE (CTCCn0-2GGAGA) is quite different from the classic GRE in terms of nucleotide sequence and the presence of a number of nucleotides between the half-sites that range from zero to two.46 In addition to GR dimer-mediated transactivation or transrepression of target genes, GR might influence gene expression independently of DNA binding. These genomic actions are mediated by protein-protein interactions of GR with several transcription factors, such as activator protein-1 (AP-1), nuclear factor-κB (NF-κB) and signal transducers and activators of transcription (STATs), thereby influencing the transcriptional activity of the latter2,4,13,18 (Figure 2).

Figure 2. Genomic, nongenomic and mitochondrial glucocorticoid signaling pathways. cPLA2α: cytosolic phospholipase A2 alpha; eNOS: endothelial nitric oxide synthetase; FKBP: immunophillins; GR: glucocorticoid receptor; HSP: heat shock proteins; MAPK: mitogen-activated protein kinases; NO: nitric oxide; PI3K: phosphatidylinositol 3-kinase; TF: transcription factor.
Besides glucocorticoid genomic actions, a growing body of evidence indicates that these hormones can induce some effects in a short time frame.47 These effects are referred to as “nongenomic”, occur in non-nucleated cells and do not require transcription/translation processes.47 Representative examples of nongenomic glucocorticoid actions are: (i) the rapid inhibition of ACTH release from the anterior lobe of the pituitary by glucocorticoid hormones;48 (ii) the immediate increased frequency of excitatory postsynaptic potentials in the hippocampus upon exposure to glucocorticoids;49 (iii) the rapid reduction in blood pressure in patients with myocardial or brain ischemia following glucocorticoid administration;50 and (iv) the rapid disruption of the T-cell receptor (TCR) complex, this accounting for some of the immunosuppressive glucocorticoid effects.51 The molecular mechanisms underlying the nongenomic glucocorticoid effects still remain an enigma in endocrine physiology and pathophysiology. Important studies have revealed a role of a membrane-bound GR that induces the activity of several kinases, such as the mitogen-activated protein kinase (MAPK) or the phosphatidylinositol 3-kinase (PI3K) pathways.52 However, the nature of this membrane-anchoring GR has not yet been clarified. Although most of the steroid receptors were shown to incorporate palmitic acid, a post-translational modification termed “S-palmitoylation” that enables membrane binding of proteins and increases protein hydrophobicity,53,54 we and others have demonstrated that GRα does not undergo S-palmitoylation, implying that other mechanisms are responsible for GRα membrane localization and mediate the nongenomic glucocorticoid actions55-57 (Figure 2).
In addition to cytoplasm, nucleus and plasma membrane, GR was also detected in mitochondria, a key intracellular organelle with pleiotropic functions including energy production, apoptosis, calcium homeostasis, thermogenesis and stress response.58 Interestingly, GREs were more than 20 years ago identified within the regulatory sites (D-loop) of the mitochondrial genome, this suggesting a genomic interrelation between mitochondria and the nucleus.59 A number of studies in several tissues have observed a cytoplasmic-to-mitochondrion GR translocation or vice versa in response to dexamethasone, indicating that mitochondrial GR is dynamically regulated upon exposure to glucocorticoids.59-61 In addition to direct GR-GRE interaction, mitochondrial gene expression is regulated indirectly by nuclear GR-GRE interactions that result in the transcription of genes encoding mitochondrial RNA-processing enzymes, nuclear respiratory factors or mitochondrial transcription factors.58 From the pharmacologic point of view, synthetic glucocorticoids are currently used in the therapeutic protocols of several hematologic malignancies due to GR-mediated apoptosis of cancer cells, a mechanism that occurs in the mitochondrion58 (Figure 2).
THE GLUCOCORTICOID SIGNALING PATHWAY IN ALLOSTASIS
In humans, any alterations in glucocorticoid signal transduction may cause short- or long-term adverse effects and affect a patient’s sense of well-being and/or performance. At the molecular level, these undesired consequences may be explained by impaired tissue sensitivity to glucocorticoids that might take the form of glucocorticoid resistance or glucocorticoid hypersensitivity, two conditions that are associated with increased morbidity.62-66
Primary Generalized Glucocorticoid
Resistance (PGGR), later named “Chrousos syndrome”, is a rare endocrinologic
condition which is characterized by partial tissue insensitivity to
glucocorticoids.63-70 Patients with this syndrome have defective
glucocorticoid negative feedback loops leading to compensatory hypersecretion
of both ACTH and AVP. The increased plasma ACTH concentrations cause
hypertrophy of the adrenal cortex and trigger the synthesis and release of
cortisol, steroid precursors with mineralocorticoid activity
(deoxy-corticosterone and corticosterone) and adrenal androgens [androstenedione,
dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS)].63-70
Depending on the functioning of these pathophysiologic mechanisms, patients may
have only biochemical alterations or present with symptoms and signs of
mineralocorticoid excess (hypertension and/or hypokalemic alkalosis) and/or
androgen excess (ambiguous genitalia at birth in karyotypic females, acne,
hirsutism, precocious puberty, male-pattern hair loss and hypofertility in both
sexes, oligo-amenorrhea and menstrual irregularities in women and oligospermia
in men).63-70 Glucocorticoid deficiency is relatively rare and has
been reported in a small number of patients, such as adults with chronic
fatigue,68,71,72 a child presenting with hypoglycemic generalized
tonic-clonic seizures that occurred during fever73 and a newborn
with hypoglycemia, manifesting of easy “fatigability” with feeding and growth
hormone deficiency.74 Some patients may also complain of profound
anxiety and depression, which may be explained by the increased secretion of ACTH
and AVP.70 The molecular basis of Chrousos syndrome has been
ascribed to inactivating point mutations, insertions or deletions in the NR3C1
gene (Table 1, Figure 3).71,73-95
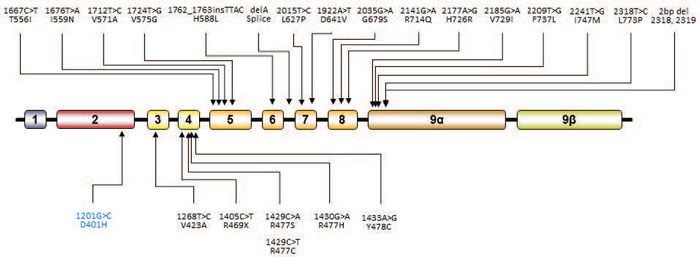
Figure 3. Schematic representation of the known mutations of the NR3C1 gene causing alterations in tissue sensitivity to glucocorticoids. Mutations indicated in dark color are associated with PGGR or Chrousos syndrome, while mutation D401H, which is indicated in blue, is the only mutation that has been associated with PGGH syndrome.
Primary Generalized Glucocorticoid Hypersensitivity (PGGH) is a rare syndrome characterized by increased tissue sensitivity to glucocorticoids mostly due to activating NR3C1 mutations or polymorphisms.63-65 Patients with this condition present with clinical manifestations of metabolic syndrome but low concentrations of serum cortisol due to compensatory hypoactivation of the HPA axis.63-65 To date, only one patient has been reported with symptoms and signs of PGGH, namely visceral obesity, type 2 diabetes, hypertension and dyslipidemia.96 The patient harbored a point heterozygous G → C substitution at nucleotide position 1201, which resulted in aspartic acid (D) to histidine (H) substitution at amino acid position 401 in the N-terminal domain of the receptor.96 Functional characterization of hGRαD401H showed that the mutant receptor increased the expression of several glucocorticoid-responsive genes.96 In addition to activating NR3C1 mutations, common polymorphisms associated with increased glucocorticoid sensitivity are N363S and BclI.18,62 Moreover, only a few cases of PGGH that do not harbor a defective NR3C1 gene have been published.97-101 The etiology of these cases has not as yet been elucidated (Figure 3).
RECENT ADVANCES IN CHROUSOS SYNDROME
We and others have systematically investigated the molecular mechanisms of action of mutant hGRs causing Chrousos syndrome by applying standard methods of molecular and structural biology (Table 1).71,73-95 Recently we functionally characterized hGRαT556I, an NR3C1 mutation identified in a 56-year old man with an adrenal incidentaloma.93 This patient was asymptomatic and had only mildly elevated morning plasma ACTH concentrations, as well as increased 24-hour urinary free cortisol excretion (UFC).89 Sequencing analysis of the NR3C1 gene revealed a novel point C→T substitution at nucleotide position 1667, which led to a threonine to isoleucine (T→I) substitution at amino acid position 556 in the LBD of the receptor.89 Our in vitro studies showed that the hGRαT556I displayed reduced ability to transactivate target genes, had decreased affinity for the ligand, demonstrated increased transrepression of the NF-κB responsive genes and required more time to translocate from the cytoplasm to nucleus following dexamethasone-induced activation.93
Velayos and collaborators recently described three patients with Chrousos syndrome caused by two novel point mutations in the NR3C1 gene.94 The first patient was a 12-year old girl who was referred to an endocrinologist because of high serum cortisol concentrations. She had mild hirsutism and a white stretch mark on the right thigh. Endocrinologic evaluation revealed high 24-hour urinary free cortisol excretion in two serial measurements, increased plasma ACTH and serum testosterone concentrations, as well as high salivary cortisol concentrations. The patient did not suppress serum cortisol concentrations following an overnight dexamethasone (1mg) suppression test. Interestingly, her mother was asymptomatic and had only slightly increased serum cortisol concentrations.94 Both the girl and her mother harbored a novel point NR3C1 mutation (1429C→T) in exon 4, which resulted in an arginine to cysteine (R→C) substitution at amino acid position 477 in the second zinc finger of the DBD of the receptor.94 The second patient was a 41-year old woman who had had a history of hirsutism for 4 years and also presented with chronic fatigue and anxiety.94 She had increased 24-hour UFC excretion and increased salivary cortisol and serum DHEA-S and testosterone concentrations. Sequencing analysis revealed a novel insertion of four bases between nucleotides 1762 and 1763, which resulted in a substitution of four amino acids at positions 588-591 leading to a premature stop codon at amino acid position 592, and therefore to a truncated protein.94
Three novel heterozygous point mutations in the NR3C1 gene have also been identified by Vitellius et al in patients with adrenal incidentalomas and increased circulating glucocorticoid concentrations without clinical manifestations suggestive of Cushing syndrome.95 Two of these mutations, R477S and Y478C, were located in the second zinc finger of the receptor DBD, whereas the third mutation, L672P, was found in the receptor LBD.95 The authors undertook a functional characterization of these three mutations. Transactivation assays showed that the mutant receptors hGRαR477S and hGRαL672P had decreased ability to transactivate glucocorticoid-responsive genes compared to the wild-type receptor (hGRαWT). Furthermore, the mutant receptors located in the DBD had a similar affinity for dexamethasone to that of hGRαWT.95 In contrast, hGRαL672P was not able to bind to the ligand because of a defective LBD. hGRαR477S and hGRαY478C had a marked delay in the translocation from the cytoplasm to the nucleus following ligand-binding, whereas hGRαL672P alone maintained its cytoplasmic localization.95 As expected, the mutant receptors hGRαR477S and hGRαY478C had reduced ability to bind to DNA compared to hGRαWT. Structural biology assays demonstrated that hGRαR477S lost two hydrogen bonds with GREs and hGRαY478C displayed impaired interaction with neighboring amino acid residues, whereas hGRαL672P caused a conformational change of helix 8.95
NOVEL INSIGHTS INTO PGGH
We have described a 9-year old girl with transient PGGH who presented with a long history of clinical manifestations suggestive of Cushing syndrome, including progressive weight gain, buffalo hump, moon facies, purple skin striae, acanthosis nigricans, hirsutism, myopathy, as well as decreased growth velocity.102 Interestingly, her morning plasma ACTH and serum cortisol concentrations were undetectable (<1 pg/mL and 0.025 mcg/dL, respectively) and remained decreased during the 24-hour study. CRH stimulation showed no increase in plasma ACTH and serum cortisol concentrations.102 These clinical manifestations and our endocrinologic investigations pointed to a diagnosis of transient generalized glucocorticoid hypersensitivity. Sequencing analysis revealed no mutations or polymorphisms in the NR3C1 gene.102 Of note, the symptoms and signs of the patient gradually resolved over the ensuing three months. RNA sequencing was performed in peripheral blood mononuclear cells during the acute and the resolution phases of the disease and transcriptomic analysis revealed 903 differentially expressed genes. The majority of them were found to be NF-κB target genes, indicating that a transient post-receptor defect or a viral or bacterial protein might have enhanced hGRα-mediated transcriptional activity.102
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Chrousos syndrome and Primary Generalized Glucocorticoid Hypersensitivity syndrome are two rare allostatic conditions which are characterized by impaired glucocorticoid signaling. The molecular basis of these conditions has been attributed to genetic defects in the NR3C1 gene. However, some patients with clinical manifestations suggestive of these syndromes did not harbor any mutations, insertions or deletions in the NR3C1 gene, implying that several other factors might influence the glucocorticoid signal transduction. Representative examples of such molecules are the FK506-Binding Immunophilins FKBPs. Indeed, any imbalance between the protein expression levels of FKBP51 and FKBP52 may cause glucocorticoid resistance or hypersensitivity.103 In addition to hGRα protein partners, accumulating evidence suggests that some of the non-coding RNAs could play an important role in determining tissue sensitivity to glucocorticoids.104 One of them, “Growth Arrest-Specific 5” (GAS5) long non-coding RNA, was found to suppress the expression of several glucocorticoid responsive genes through binding to the DBD of GRα by acting as a decoy GRE.105 GAS5 was identified as an important factor influencing human response to glucocorticoids. Lucafo et al demonstrated that poor responders to glucocorticoids had increased levels of GAS5 compared to good responders.106 These accumulating data evidence the fact that novel technologies, including whole-genome sequencing, whole-exome sequencing and RNA sequencing, will undoubtedly enable us to acquire a deeper understanding of the GR “interactome”.
FUNDING
This work was supported by the European Union (European Social Fund – ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) – Research Funding Program: THALIS - University of Athens (UOA), Athens, Greece.
REFERENCES
1. Charmandari E, Tsigos C, Chrousos GP, 2005 Endocrinology of the stress response. Annu Rev Physiol 67: 259-284.
2. Chrousos GP, Kino T, 2005 Intracellular glucocorticoid signaling: A formerly simple system turns stochastic. Sci STKE 304: pe48.
3. Chrousos GP, Kino T, 2007 Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress 10: 213-219.
4. Nicolaides NC, Kyratzi E, Lamprokostopoulou A, Chrousos GP, Charmandari E, 2015 Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 22: 6-19.
5. Nicolaides NC, Charmandari E, Chrousos GP, 2014 Adrenal Steroid Hormone Secretion: Physiologic and Endocrine Aspects. Elsevier Reference Module in Biomedical Sciences.
6. Talaber G, Jondal M, Okret S, 2013 Extra-adrenal glucocorticoid synthesis: immune regulation and aspects on local organ homeostasis. Mol Cell Endocrinol 380: 89-98.
7. Nicolaides NC, Charmandari E, Chrousos GP, Kino T, 2014 Circadian endocrine rhythms: the hypothalamic-pituitary-adrenal axis and its actions. Ann N Y Acad Sci 1318: 71-80.
8. Nicolaides NC, Charmandari E, Kino T, Chrousos GP, 2017 Stress-Related and Circadian Secretion and Target Tissue Actions of Glucocorticoids: Impact on Health. Front Endocrinol 8: 70.
9. Waite EJ, McKenna M, Kershaw Y, et al, 2012 Ultradian corticosterone secretion is maintained in the absence of circadian cues. Eur J Neurosci 36: 3142-3150.
10. Kalsbeek A, Palm IF, La Fleur SE, et al, 2006 SCN outputs and the hypothalamic balance of life. J Biol Rhythms 21: 458-469.
11. Leliavski A, Dumbell R, Ott V, Oster H, 2015 Adrenal clocks and the role of adrenal hormones in the regulation of circadian physiology. J Biol Rhythms 30: 20-34.
12. Zannas AS, Chrousos GP, 2017 Epigenetic programming by stress and glucocorticoids along the human lifespan. Mol Psychiatry 22: 640-646.
13. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E, 2010 The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 75: 1-12.
14. Galon J, Franchimont D, Hiroi N, et al, 2002 Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J 16: 61-71.
15. Rhen T, Cidlowski JA, 2005 Anti-inflammatory action of glucocorticoids--new mechanisms for old drugs. N Engl J Med 353: 1711-1723.
16. Oakley RH, Cidlowski JA, 2011 Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286: 3177-3184.
17. Oakley RH, Cidlowski JA, 2013 The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132: 1033-1044.
18. Ramamoorthy S, Cidlowski JA, 2016 Corticosteroids: Mechanisms of Action in Health and Disease. Rheum Dis Clin North Am 42: 15-31.
19. Bamberger CM, Bamberger AM, de Castro M, Chrousos GP, 1995 Glucocorticoid receptor β, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 95: 2435-2441.
20. Charmandari E, Chrousos GP, Ichijo T, et al, 2005 The human glucocorticoid receptor (hGR) β isoform suppresses the transcriptional activity of hGRα by interfering with formation of active coactivator complexes. Mol Endocrinol 19: 52-64.
21. Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA, 2003 Molecular origins for the dominant negative function of human glucocorticoid receptor β. Mol Cell Biol 23: 4319-4330.
22. Kelly A, Bowen H, Jee YK, et al, 2008 The glucocorticoid receptor beta isoform can mediate transcriptional repression by recruiting histone deacetylases. Allergy Clin Immunol 121: 203-208.
23. Kim SH, Kim DH, Lavender P, et al, 2009 Repression of TNF-alpha-induced IL-8 expression by the glucocorticoid receptor-beta involves inhibition of histone H4 acetylation. Exp Mol Med 41: 297-306.
24. Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP, 2009 Glucocorticoid receptor (GR) β has intrinsic, GRα-independent transcriptional activity. Biochem Biophys Res Commun 381: 671-675.
25. Kino T, Su YA, Chrousos GP, 2009 Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci 66: 3435-3448.
26. Chatzopoulou A, Schoonheim PJ, Torraca V, Meijer AH, Spaink HP, Schaaf MJ, 2017 Functional analysis reveals no transcriptional role for the glucocorticoid receptor β-isoform in zebrafish. Mol Cell Endocrinol 447: 61-70.
27. Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, Hinds TD Jr 2014 Glucocorticoid receptor β stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem 289: 17885-17894.
28. He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA, 2015 Human Glucocorticoid Receptor β Regulates Gluconeogenesis and Inflammation in Mouse Liver. Mol Cell Biol 36: 714-730.
29. Yin Y, Zhang X, Li Z, et al, 2013 Glucocorticoid receptor β regulates injury-mediated astrocyte activation and contributes to glioma pathogenesis via modulation of β-catenin/TCF transcriptional activity. Neurobiol Dis 59: 165-176.
30. Wang Q, Lu PH, Shi ZF, et al, 2015 Glucocorticoid Receptor β acts as a co-activator of T-cell factor 4 and enhances glioma cell proliferation. Mol Neurobiol 52: 1106-1118.
31. McBeth L, Nwaneri AC, Grabnar M, Demeter J, Nestor-Kalinoski A, Hinds TD Jr, 2016 Glucocorticoid receptor beta increases migration of human bladder cancer cells. Oncotarget 7: 27313-27324.
32. Lewis-Tuffin LJ, Cidlowski JA, 2006 The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci 1069: 1-9.
33. Ray DW, Davis JR, White A, Clark AJ, 1996 Glucocorticoid receptor structure and function in glucocorticoid-resistant small cell lung carcinoma cells. Cancer Res 56: 3276-3280.
34. Beger C, Gerdes K, Lauten M, et al, 2003 Expression and structural analysis of glucocorticoid receptor isoform gamma in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. Br J Haematol 122: 245-252.
35. Rivers C, Levy A, Hancock J, Lightman S, Norman M, 1999 Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. J Clin Endocrinol Metab 84: 4283-4286.
36. Krett NL, Pillay S, Moalli PA, Greipp PR, Rosen ST, 1995 A variant glucocorticoid receptor messenger RNA is expressed in multiple myeloma patients. Cancer Res 55: 2727-2729.
37. de Lange P, Segeren CM, Koper JW, et al, 2001 Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Res 61: 3937-3941.
38. Gaitan D, DeBold CR, Turney MK, Zhou P, Orth DN, Kovacs WJ, 1995 Glucocorticoid receptor structure and function in an adrenocorticotropin-secreting small cell lung cancer. Mol Endocrinol 9: 1193-1201.
39. Lu NZ, Cidlowski JA, 2005 Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell 18: 331-342.
40. Lu NZ, Collins JB, Grissom SF, Cidlowski JA, 2007 Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol Cell Biol 27: 7143-7160.
41. Wu I, Shin SC, Cao Y, et al, 2013 Selective glucocorticoid receptor translational isoforms reveal glucocorticoid-induced apoptotic transcriptomes. Cell Death Dis 4: e453.
42. Grad I, Picard D, 2007 The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol 275: 2-12.
43. Pratt WB, Toft DO, 1997 Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev 18: 306-360.
44. Beato M, 1989 Gene regulation by steroid hormones. Cell 56: 335-344.
45. Surjit M, Ganti KP, Mukherji A, et al, 2011 Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145: 224-241.
46. Hudson WH, Youn C, Ortlund EA, 2013 The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol 20: 53-58.
47. Groeneweg FL, Karst H, de Kloet ER, Joëls M, 2012 Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol 350: 299-309.
48. Hinz B, Hirschelmann R, 2000 Rapid non-genomic feedback effects of glucocorticoids on CRF-induced ACTH secretion in rats. Pharm Res 17: 1273-1277.
49. Karst H, Berger S, Turiault M, Tronche F, Schütz G, Joëls M, 2005 Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA 102: 19204-19207.
50. Hafezi-Moghadam A, Simoncini T, Yang Z, et al, 2002 Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med 8: 473-479.
51. Löwenberg M, Verhaar AP, Bilderbeek J, et al, 2006 Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep 7: 1023-1029.
52. Samarasinghe RA, Witchell SF, DeFranco DB, 2012 Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle 11: 2819-2827.
53. Marino M, Ascenzi P, Acconcia F, 2006 S-palmitoylation modulates estrogen receptor alpha localization and functions. Steroids 71: 298-303.
54. Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER, 2007 A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem 282: 22278-22288.
55. Nicolaides NC, Kino T, Roberts ML, et al, 2017 The Role of S-palmitoylation of the human glucocorticoid receptor (hGR) in mediating the nongenomic glucocorticoid actions. J Mol Biochem [In press].
56. Samarasinghe RA, Di Maio R, Volonte D, et al, 2011 Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci U S A 108: 16657-16662.
57. Deng Q, Waxse B, Riquelme D, Zhang J, Aguilera G, 2015 Helix 8 of the ligand binding domain of the glucocorticoid receptor (GR) is essential for ligand binding. Mol Cell Endocrinol 408: 23-32.
58. Lee SR, Kim HK, Song IS, et al, 2013 Glucocorticoids and their receptors: insights into specific roles in mitochondria. Prog Biophys Mol Biol 112: 44-54.
59. Demonacos C, Djordjevic-Markovic R, Tsawdaroglou N, Sekeris CE, 1995 The mitochondrion as a primary site of action of glucocorticoids: the interaction of the glucocorticoid receptor with mitochondrial DNA sequences showing partial similarity to the nuclear glucocorticoid responsive elements. J Steroid Biochem Mol Biol 55: 43-55.
60. Demonacos C, Tsawdaroglou NC, Djordjevic-Markovic R, et al, 1993 Import of the glucocorticoid receptor into rat liver mitochondria in vivo and in vitro. J Steroid Biochem Mol Biol 46: 401-413.
61. Psarra AM, Sekeris CE, 2011 Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta 1813: 1814-1821.
62. Quax RA, Manenschijn L, Koper JW, et al, 2013 Glucocorticoid sensitivity in health and disease. Nat Rev Endocrinol 9: 670-686.
63. Charmandari E, 2011 Primary generalized glucocorticoid resistance and hypersensitivity. Horm Res Paediatr 76: 145-155.
64. Charmandari E, 2012 Primary generalized glucocorticoid resistance and hypersensitivity: the end-organ involvement in the stress response. Sci Signal 5: pt5.
65. Charmandari E, Kino T, Chrousos GP, 2013 Primary generalized familial and sporadic glucocorticoid resistance (Chrousos syndrome) and hypersensitivity. Endocr Dev 24: 67-85.
66. Nicolaides NC, Charmandari E, Chrousos GP, Kino T, 2014 Recent advances in the molecular mechanisms determining tissue sensitivity to glucocorticoids: novel mutations, circadian rhythm and ligand-induced repression of the human glucocorticoid receptor. BMC Endocr Disord 14: 71.
67. Charmandari E, Kino T, 2010 Chrousos syndrome: a seminal report, a phylogenetic enigma and the clinical implications of glucocorticoid signaling changes. Eur J Clin Invest 40: 932-942.
68. Charmandari E, Kino T, Ichijo T, Chrousos GP, 2008 Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab 93: 1563-1572.
69. Chrousos G, 2011 Q&A: primary generalized glucocorticoid resistance. BMC Med 9: 27.
70. Nicolaides NC, Charmandari E, 2015 Chrousos syndrome: from molecular pathogenesis to therapeutic management. Eur J Clin Invest 45: 504-514.
71. Chrousos GP, Vingerhoeds A, Brandon D, et al, 1982 Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest 69: 1261-1269.
72. Chrousos GP, Detera-Wadleigh SD, Karl M, 1993 Syndromes of glucocorticoid resistance. Ann Intern Med 119: 1113-1124.
73. Nader N, Bachrach BE, Hurt DE, et al, 2010 A novel point mutation in the helix 10 of the human glucocorticoid receptor causes generalized glucocorticoid resistance by disrupting the structure of the ligand-binding domain. J Clin Endocrinol Metab 95: 2281-2285.
74. McMahon SK, Pretorius CJ, Ungerer JP, et al, 2010 Neonatal complete generalized glucocorticoid resistance and growth hormone deficiency caused by a novel homozygous mutation in Helix 12 of the ligand binding domain of the glucocorticoid receptor gene (NR3C1). J Clin Endocrinol Metab 95: 297-302.
75. Nicolaides N, Lamprokostopoulou A, Sertedaki A, Charmandari E, 2016 Recent advances in the molecular mechanisms causing primary generalized glucocorticoid resistance. Hormones (Athens) 15: 23-34.
76. Karl M, Lamberts SW, Koper JW, et al, 1996 Cushing’s disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Physicians 108: 296-307.
77. Hurley DM, Accili D, Stratakis CA, et al, 1991 Point mutation causing a single amino acid substitution in the hormone binding domain of the glucocorticoid receptor in familial glucocorticoid resistance. J Clin Invest 87: 680-686.
78. Karl M, Lamberts SW, Detera-Wadleigh SD, et al, 1993 Familial glucocorticoid resistance caused by a splice site deletion in the human glucocorticoid receptor gene. J Clin Endocrinol Metab 76: 683-689.
79. Malchoff DM, Brufsky A, Reardon G, et al, 1993 A mutation of the glucocorticoid receptor in primary cortisol resistance. J Clin Invest 91: 1918-1925.
80. Kino T, Stauber RH, Resau JH, Pavlakis GN, Chrousos GP, 2001 Pathologic human GR mutant has a transdominant negative effect on the wild-type GR by inhibiting its translocation into the nucleus: importance of the ligand-binding domain for intracellular GR trafficking. J Clin Endocrinol Metab 86: 5600-5608.
81. Ruiz M, Lind U, Gafvels M, et al, 2001 Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol (Oxf) 55: 363-371.
82. Mendonca BB, Leite MV, de Castro M, et al, 2002 Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab 87: 1805-1809.
83. Vottero A, Kino T, Combe H, Lecomte P, Chrousos GP, 2002 A novel, C-terminal dominant negative mutation of the GR causes familial glucocorticoid resistance through abnormal interactions with p160 steroid receptor coactivators. J Clin Endocrinol Metab 87: 2658-2667.
84. Charmandari E, Kino T, Vottero A, Souvatzoglou E, Bhattacharyya N, Chrousos GP, 2004 Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: Molecular genotype, genetic transmission and clinical phenotype. J Clin Endocrinol Metab 89: 1939-1949.
85. Charmandari E, Raji A, Kino T, et al, 2005 A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of hGR LBD in conferring transactivational activity. J Clin Endocrinol Metab 90: 3696-3705.
86. Charmandari E, Kino T, Ichijo T, Zachman K, Alatsatianos A, Chrousos GP, 2006 Functional characterization of the natural human glucocorticoid receptor (hGR) mutants hGRαR477H and hGRαG679S associated with generalized glucocorticoid resistance. J Clin Endocrinol Metab 91: 1535-1543.
87. Charmandari E, Kino T, Ichijo T, et al, 2007 A novel point mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J Clin Endocrinol Metab 92: 3986-3990.
88. Bouligand J, Delemer B, Hecart AC, et al, 2010 Familial glucocorticoid receptor haploinsufficiency by non-sense mediated mRNA decay, adrenal hyperplasia and apparent mineralocorticoid excess. PLoS One 5: e13563.
89. Zhu HJ, Dai YF, Wang O, et al, 2011 Generalized glucocorticoid resistance accompanied with an adrenocortical adenoma and caused by a novel point mutation of human glucocorticoid receptor gene. Chin Med J (Engl) 124: 551-555.
90. Roberts ML, Kino T, Nicolaides NC, et al, 2013 A novel point mutation in the DNA-binding domain (DBD) of the human glucocorticoid receptor causes primary generalized glucocorticoid resistance by disrupting the hydrophobic structure of its DBD. J Clin Endocrinol Metab 98: E790-E795.
91. Nicolaides NC, Roberts ML, Kino T, et al, 2014 A novel point mutation of the human glucocorticoid receptor gene causes primary generalized glucocorticoid resistance through impaired interaction with the LXXLL motif of the p160 coactivators: dissociation of the transactivating and transreppressive activities. J Clin Endocrinol Metab 99: E902-E907.
92. Nicolaides NC, Geer EB, Vlachakis D, et al, 2015 A novel mutation of the hGR gene causing Chrousos syndrome. Eur J Clin Invest 45: 782-791.
93. Nicolaides NC, Skyrla E, Vlachakis D, et al, 2016 Functional characterization of the hGRαT556I causing Chrousos syndrome. Eur J Clin Invest 46: 42-49.
94. Velayos T, Grau G, Rica I, Pérez-Nanclares G, Gaztambide S, 2016 Glucocorticoid resistance syndrome caused by two novel mutations in the NR3C1 gene. Endocrinol Nutr 63: 369-371.
95. Vitellius G, Fagart J, Delemer B, et al, 2016 Three novel heterozygous point mutations of NR3C1 causing glucocorticoid resistance. Hum Mutat 37: 794-803.
96. Charmandari E, Ichijo T, Jubiz W, et al, 2008 A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hGR) gene enhancing hGR-mediated gene expression. J Clin Endocrinol Metab 93: 4963-4968.
97. Iida S, Nakamura Y, Fujii H, et al, 1990 A patient with hypocortisolism and Cushing’s syndrome-like manifestations: cortisol hyperreactive syndrome. J Clin Endocrinol Metab 70: 729-737.
98. Fujii H, Iida S, Gomi M, Tsugawa M, Kitani T, Moriwaki K, 1993 Augmented induction by dexamethasone of metallothionein IIa messenger ribonucleic acid in fibroblasts from a patient with cortisol hyperreactive syndrome. J Clin Endocrinol Metab 76: 445-449.
99. Newfield RS, Kalaitzoglou G, Licholai T, et al, 2000 Normocortisolemic Cushing’s syndrome initially presenting with increased glucocorticoid receptor numbers. J Clin Endocrinol Metab 85: 14-21.
100. Russcher H, Smit P, van Rossum EF, et al, 2006 Strategies for the characterization of disorders in cortisol sensitivity. J Clin Endocrinol Metab 91: 694-701.
101. Krysiak R, Okopien B, 2012 Glucocorticoid hypersensitivity syndrome-a case report. West Indian Med J 61: 844-846.
102. Nicolaides NC, Lamprokostopoulou A, Polyzos A, et al, 2015 Transient generalized glucocorticoid hypersensitivity. Eur J Clin Invest 45: 1306-1315.
103. Wochnik GM, Ruegg J, Abel GA, Schmidt U, Holsboer F, Rein T, 2005 FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem 280: 4609-4616.
104. Wang H, Gou X, Jiang T, Ouyang J, 2017 The effects of microRNAs on glucocorticoid responsiveness. J Cancer Res Clin Oncol 143: 1005-1011.
105. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP, 2010 Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal 3: ra8.
106. Lucafo M, De Iudicibus S, Di Silvestre A, et al, 2015 Long noncoding RNA GAS5: a novel marker involved in glucocorticoid response. Curr Mol Med 15: 94-99.
Address for correspondence:
Nicolas C. Nicolaides, MD, PhD. Division of Endocrinology and Metabolism, Center of Clinical, Experimental Surgery and Translational Research, Biomedical Research Foundation of the Academy of Athens, 4 Soranou tou Efessiou Street, Athens, 11527, Greece; Tel.: +30-210-7487314, Fax: +30-210-6597545, E-mail: nnicolaides@bioacademy.gr
Received: 26-05-2017, Accepted: 31-05-2017