1Institute of Endocrinology and Metabolism, Rabin Medical Center, Beilinson Hospital, Petah Tiqva; Sackler School of Medicine, Tel Aviv University, Israel; 2Institute of Neuropathology, Hamburg University, Germany; 3Department of Internal Medicine and Endocrine Unit, Medical School and Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
OBJECTIVE: To present two female patients with
acromegaly inadequately controlled with long-acting octreotide who were
subsequently treated with the multireceptor-targeted somatostatin analogue
pasireotide that over-suppressed IGF-1 levels.
METHODS: We report two patients
who failed surgery and received long-acting octreotide 20-30 mg/month as part
of two double-blind, Phase III clinical trials. After 6-12 months of octreotide
treatment, both patients remained inadequately controlled and were switched to
long-acting pasireotide 40 mg/month as part of a crossover extension phase.
RESULTS: During the core phase of the studies the patients received octreotide
20-30 mg/month, but GH and IGF-1 levels remained above normal. They were
switched to pasireotide 40 mg/month after 6 and 12 months, according to the
study protocols. After crossover, GH and IGF-1 decreased and normalized, but
continued treatment led to further reduction of IGF-1 to below the normal;
these reduced levels mildly increased following pasireotide dose reduction to
20 mg/month. Tumour volume was reduced and the clinical signs and symptoms of
acromegaly also improved.
CONCLUSION: These patients achieved long-term
biochemical control, tumour volume reduction and improvement of clinical
signs/symptoms after switching from octreotide to pasireotide. IGF-1
over-suppression is observed in a few patients and requires dose adjustment of
pasireotide.
Acromegaly, Growth hormone, Insulin-like growth factor 1, Octreotide, Pasireotide
INTRODUCTION
Acromegaly is a rare, serious, multisystem disease resulting from increased growth hormone (GH) secretion due in most cases to a pituitary adenoma,1 with subsequent increases in insulin-like growth factor 1 (IGF-1) levels. Patients with active disease have higher morbidity and mortality than the general population.2 The goals of treatment are to control the levels of both GH and IGF-1, reduce and/or stabilize tumour size, preserve pituitary function and prevent recurrence.3
Transsphenoidal surgery is generally recommended as first-line therapy.4 However, even in referral centres, approximately 50% of the patients will experience persistent or recurrent disease and require medical treatment to achieve disease control.5 Long-acting somatostatin analogues are the cornerstone of medical therapy for acromegaly and are indicated in patients with failed surgery or as first-line treatment when surgery is contraindicated or declined.4 However, many patients do not achieve biochemical control with the somatostatin receptor subtype sst-2 preferential analogues octreotide and lanreotide.6-8
Pasireotide is a multireceptor-targeted somatostatin analogue with high affinity for sst2, sst3 and sst5.9 In a large randomized Phase III study in patients with medically naïve acromegaly, long-acting pasireotide was significantly superior to long-acting octreotide in providing biochemical control at month 12 (a head to-head superiority study).10 In a second randomized Phase III study evaluating long-acting pasireotide versus long-acting octreotide or lanreotide autogel in somatostatin analogue-resistant patients with acromegaly (PAOLA study), pasireotide provided superior efficacy compared with continued treatment with octreotide or lanreotide for 24 weeks.11 We report our experience with two patients enrolled in these studies who switched from octreotide to pasireotide during a crossover extension phase and who over-responded to pasireotide. The sst profile of these two somatotropinomas was analyzed.
METHODS
Serum GH and IGF-1 in the head to-head study (patient 1) were measured using chemiluminescent immunometric assays [Immulite 2000/1000; Diagnostic Products Corp (Siemens)]; samples were analyzed by Quest Diagnostics Nichols Institute Laboratory between March 2008 and March 2010 and then by Quest Diagnostics Clinical Trials Laboratory. Growth hormone and IGF-1concentrations in the PAOLA study (patient 2) were also analyzed by Quest Diagnostic Clinical Trials using chemiluminescent immunometric assays [Immulite 2000; Diagnostic Products Corp (Siemens)].
Patient 1 adenoma was immunostained for the different ssts using antibodies against sst2a (Zytomed; dilution, 1:150), sst3 (provided by S. Schulz, Jena, Germany; dilution, 1:10) and sst5 (Zytomed; dilution, 1:150). The sst 1, 2, 3 and 5 mRNA expression of patient 2 adenoma was measured by real-time RT-PCR and compared with the median of 23 other GH-adenomas evaluated at the same time, as previously published.12
CASE 1
In November 2008, a 59-year-old female presented with characteristic features of acromegaly including enlarged face and hands, macroglossia, snoring and operations for bilateral carpal tunnel syndrome. She had previously undergone thyroid surgery for retrosternal goitre and resection of the parathyroid gland for primary hyperparathyroidism with severe hypercalcaemia. She was treated with thyroid hormone replacement therapy (100 µg/day) and metformin (850 mg/day) for type 2 diabetes mellitus. Genetic studies were negative for multiple endocrine neoplasia types 1 and 4.
Hormonal evaluation showed elevated levels of serum GH (>40 µg/L), IGF-1 (124-127 nmol/L; normal, 10.5-29.2), parathyroid hormone (98 ng/L; normal, 10-65) and serum calcium (10.9-11.2 mg/dL; normal, 8.5-10.5). Prolactin (PRL) levels were normal. Sellar magnetic resonance imaging (MRI) demonstrated a left pituitary macroadenoma (15×11 mm) with left cavernous sinus involvement but without suprasellar extension. A diagnosis of GH-secreting adenoma was suspected. In June 2009, the patient underwent transsphenoidal surgery and the adenoma was partially resected; immunostaining of the tumour showed both GH and PRL expression. Post-operative hormonal evaluation showed active acromegaly with elevated GH (16-21 µg/L) and IGF-1 (73 nmol/L) levels.
Medical therapy with a somatostatin analogue was offered within the Phase III clinical trial of long-acting pasireotide versus long-acting octreotide.10 The patient was randomized to receive octreotide 20 mg/month in August 2009. At baseline, a pituitary tumour remnant of 11×11 mm was visible by MRI in the left side of the sella, with minimal cavernous sinus invasion (Figure 1A). Her study baseline GH (mean of five consecutive measurements every 30 minutes) and IGF-1 levels were 9.5 µg/L and 96.6 nmol/L (normal, 12-24.7), respectively. After 3 months, mean GH and IGF-1 levels decreased to 3.3 µg/L and 65.1 nmol/L, respectively (Figure 2). The octreotide dose was increased to 30 mg/month, but mean GH (3.6 µg/L) and IGF-1 (63.4 nmol/L) levels remained elevated at 12 months. Tumour volume was reduced, measuring 10×4.5 mm at the end of 12 months (Figure 1B and 1C). During octreotide therapy, treatment with metformin 850 mg BID provided glycaemic control (HbA1c, 6.0-6.2%, normal, 4-5.9%).
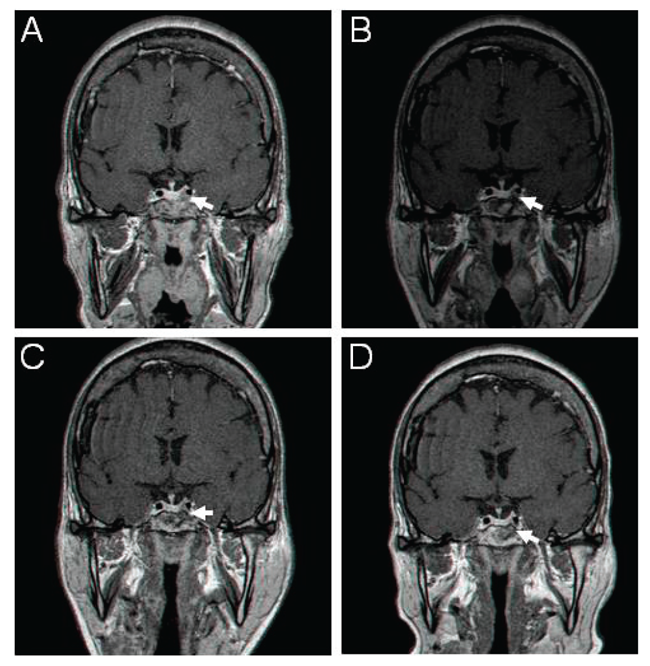
Figure 1. Sellar gadolinium-enhanced MR images at baseline and during treatment with long-acting octreotide and pasireotide in patient 1. (A) prior to octreotide treatment; (B) after 6 months of treatment with octreotide; (C) after 12 months of treatment with octreotide; and (D) after 12 months of treatment with pasireotide. Arrows show the left-sided adenoma.
In August 2010, after 12 months the patient was switched to pasireotide, because of inadequate biochemical control as per study protocol, and was started on treatment with pasireotide 40 mg/month. After 3 months of treatment, GH and IGF-1 levels decreased to 0.8 µg/L and 9.6 nmol/L, respectively. The patient continued with the same pasireotide dose and IGF-1 levels were maintained below the lower limit of normal (LLN; Figure 2). After 12 months of pasireotide treatment, MRI revealed further tumour volume reduction (5×4.5 mm; Figure 1D). Following the initiation of pasireotide treatment, HbA1c levels increased to 7.5% after 3 months. The addition of sitagliptin (100 mg/day) and glimepiride (1 mg/day) to metformin therapy resulted in a decrease in HbA1C levels to 6.4-6.6%.
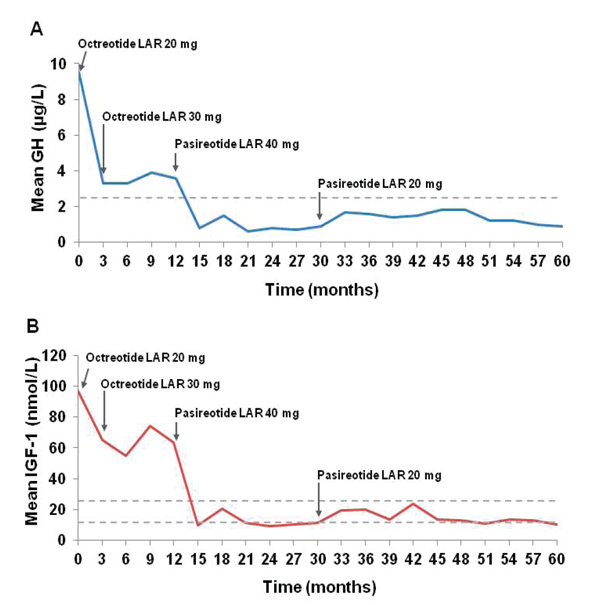
Figure 2. Switching to pasireotide suppressed (A) GH and (B) IGF-1 levels in patient 1. The patient was randomized to receive long-acting octreotide 20 mg. Following 3 months of treatment, the dose was increased to 30 mg. After 12 months the patient was switched to pasireotide (40 mg/month). A dose reduction to 20 mg occurred at month 30. The dashed lines represent the ULN for GH (2.5 µg/L) and the controlled range for IGF-1 (12-24.7 nmol/L).
After 18 months of pasireotide treatment, the dose was decreased to 20 mg/month. GH increased slightly from 0.6-0.9 µg/L to 1.4-1.7 µg/L, while IGF-1 levels increased from <LLN to within the normal range (Figure 2). The patient continued to receive pasireotide 20 mg/month for a further 40 months. Long-term treatment with pasireotide also led to improvements in enlarged face and hands and snoring. At the last follow-up (month 70), MRI showed only a small tumour remnant and laboratory analysis indicated control of GH and IGF-1 levels, 07-0.8 µg/L and 11.4-15.3 nmol/L, respectively. Serum calcium was 10.9 mg/dL. HbA1c levels were well controlled (5.8%) with metformin 1700 mg/day, sitagliptin 100 mg/day and glimepiride 2 mg/day. The patient did not experience any other adverse events during treatment.
Immunostaining for Somatostatin receptors
The adenoma was immunostained for antibodies against sst2a, sst3 and sst5 and showed moderate membranous expression of somatostatin receptors sst2a and sst5 (Figure 3) and no sst3 expression.

Figure 3. Patient 1 adenoma immunostained for sst2a, sst3 and sst5. Moderate membranous expression of sst2a and sst5 was recorded, with no sst3 expression (not shown).
CASE 2
In 2008, a 28-year old female presented with hands and feet enlargement, hyperhidrosis, polyarthralgia, paresthesia, headache, snoring and characteristic acromegalic appearance.
Hormonal evaluation showed elevated baseline and nadir GH on oral glucose tolerance test (53.9 and 34.2 µg/L, respectively) and high IGF-1 (968 µg/L, normal 117-321) (Figure 4). Prolactin, free T4 and cortisol levels were normal. Glucose was 85 mg/dL. MRI depicted a right intra and suprasellar mass with 21×17×19 mm (Figure 5A).
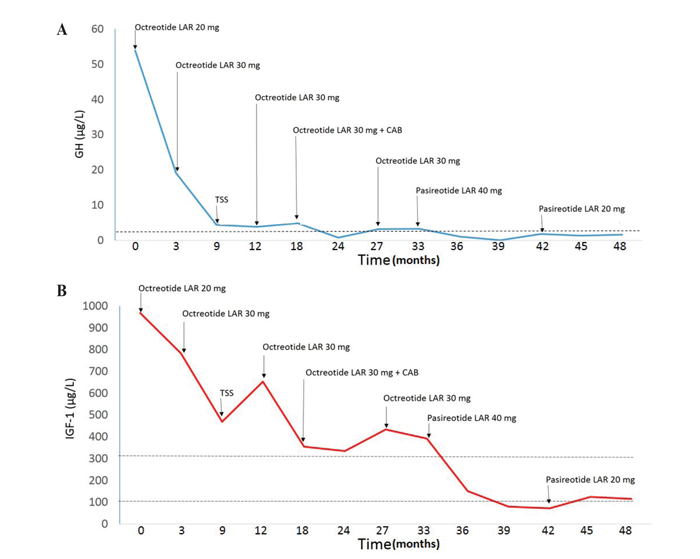
Figure 4. Pasireotide suppressed GH (A) and IGF-1 (B) in patient 2 resistant to multiple treatment regimens. The patient treated with long-acting octreotide, underwent transsphenoidal surgery (TSS) and was given combination therapy with octreotide and cabergoline (CAB) without biochemical control. After switching to pasireotide 40 mg/month, IGF-1 decreased to levels below the LLN, which resolved after dose decrease to 20 mg. The dashed lines represent the ULN for GH and the controlled range for IGF-1 during the PAOLA study.
The patient started primary medical treatment with long-acting octreotide 20 mg with only partial response after 3 months; GH and IGF-1 levels decreased to 19.1 µg/L and 782 µg/L, respectively. The dose was increased to 30 mg and after 6 months the GH and IGF-1 were still elevated, 4.4 µg/L and 469 µg/L, respectively (Figure 4).
Transsphenoidal surgery was performed, which resulted in partial resection without hormonal remission (GH, 3.84 µg/L and IGF-1, 653 µg/L) (Figure 4). MRI showed a right-sided tumour remnant (Figure 5B). Octreotide 30 mg was reintroduced and after 6 months GH and IGF-1 were 4.9 µg/L and 355 µg/L (normal, 87-238). Cabergoline (up to 2.5 mg/week) was added with partial control (GH, 0.8 µg/L; IGF-1, 334 µg/L) (Figure 4).
The patient was enrolled in a Phase III clinical trial (PAOLA study) evaluating pasireotide long-acting versus long-acting octreotide or lanreotide Autogel in somatostatin analogue-resistant patients.11 At baseline, GH and IGF-1 levels were 3.2 µg/L and 434 µg/L (normal, 115-307), respectively. During the core phase, the patient was assigned to continue treatment with octreotide for 6 months with no change in GH and IGF-1 levels (GH, 3.3 µg/L and IGF-1, 392 µg/L). During the extension phase, the patient was allowed to switch to pasireotide 40 mg as per protocol and after 3 months GH and IGF-1 were normalized (GH, 1.1 µg/L; IGF-1, 151 µg/L). Continued treatment led to a further reduction in IGF-1 levels to below the LLN (80 and 72 µg/L), so pasireotide dose was reduced to 20 mg with normalization of IGF-1 levels (125 and 115 µg/L) (Figure 4). Tumour volume was reduced by approximately 30% during pasireotide treatment (Figure 5D). Before pasireotide treatment, glucose and HbA1c levels were 93 mg/dL and 5.1%, respectively. There was a mild increase in these parameters to 111 mg/dL and 6.2%, respectively, after 6 months of pasireotide treatment. At this time, the patient was oriented with respect to lifestyle changes (diet and exercises) and improved HbA1c to 5.7%.
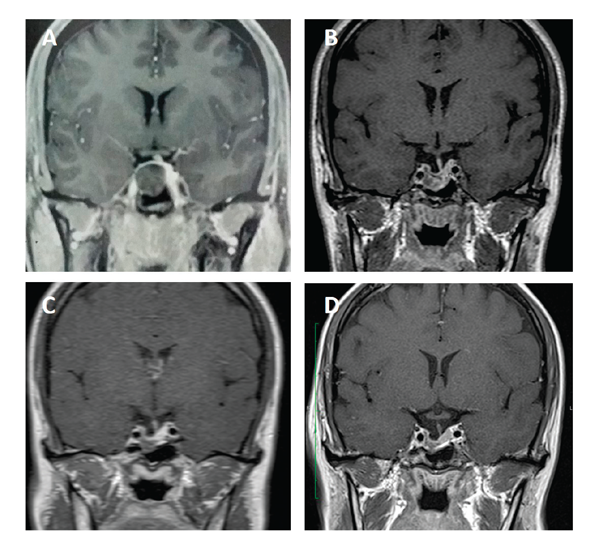
Figure 5. Sellar gadolinium-enhanced MR coronal images at diagnosis and during treatment with long-acting octreotide and pasireotide in patient 2. (A) at diagnosis; (B) 3 months after surgery; (C) after 9 months of treatment with octreotide and cabergoline; and (D) after 18 months of treatment with pasireotide.
Somatostatin receptors expression
Somatostatin receptors mRNA expression was evaluated by real-time RT-PCR and compared with the median of 23 other GH-adenomas evaluated at the same time. sst1, 2, 3 and 5 were 4.7, 1.1, 15.8 and 2.8 times the median of the 23 somatotropinomas cohort (Table 1).
DISCUSSION
Acromegaly is a rare disease with an estimated prevalence of 40-124 cases/million and is associated with increased morbidity and mortality.13 Normalization of GH and IGF-1 can restore mortality levels to that of the general population2 Somatostatin analogues have been shown to control GH and IGF-1, achieve tumour volume reduction and improve cardiovascular comorbidities in these patients.8,14 However, many patients fail to achieve biochemical control with somatostatin analogues.15 Pegvisomant is useful in patients who are inadequately controlled by somatostatin analogues, either as monotherapy or in combination. Although it has been shown to normalize IGF-1 levels in 63% of patients, pegvisomant has no effect on pituitary tumour volume.16
Here we report two unique patients with acromegaly who were uncontrolled with octreotide and, when switched to pasireotide, suppressed IGF-1 to below the LLN and required reduction of pasireotide to the minimal dose. Patient 1 was one of 182 patients naïve to medical treatment (80 of whom had undergone prior surgery) who received long-acting octreotide in the 12-month core phase of a randomized double-blind clinical trial. During the core study, long-acting pasireotide patients were more likely than octreotide patients to achieve biochemical control at month 12, with a higher rate of IGF-1 normalization (38.6% versus 23.6%; P=0.002).10 This difference could be in part due to pasireotide having a higher affinity for sst5 than octreotide. Following treatment with octreotide and despite up-titration to 30 mg, this patient did not achieve biochemical control. The patient was one of 81 patients who subsequently switched to pasireotide at month 12, and 17 (21%) of these patients achieved biochemical control after switching to pasireotide, whereas only one (2.6%) of the 38 patients switching to octreotide LAR achieved biochemical control.17 Following crossover to pasireotide, the patient’s IGF-1 level was below the LLN and led to dose reduction. Long-term treatment with the reduced dose resulted in IGF-1 levels just above the LLN.
A recent 24-week Phase III study has evaluated long-acting pasireotide versus continued treatment with long-acting octreotide or lanreotide Autogel (active control) in patients with inadequately controlled acromegaly.11 More patients treated with pasireotide achieved biochemical control (15.4% and 20.0% vs. 0%; P=0.0006 and P<0.0001, respectively) and tumour volume reduction >25% (18.5% and 10.8% vs. 1.5%) versus active control at 24 weeks.11 Patient 2 was one of the 68 patients randomly assigned to continue treatment with long-acting octreotide or lanreotide Autogel for 24 weeks. The patient did not achieve biochemical control and was one of the 50 uncontrolled patients switched to pasireotide 40 mg as per study protocol. At extension week 28, 20% of patients in the crossover group had biochemical control, some after dose escalation to 60 mg.18 Following crossover to pasireotide, the patient’s IGF-1 level was below the LLN, similarly to patient 1, and required a dose reduction to 20 mg that still resulted in low IGF-1 levels, just above the LLN.
The pituitary adenomas resected during surgery were studied for the expression of ssts. In tumour 1 moderate staining for sst2 and sst5 was recorded (Figure 3). For tumour 2 the mRNA expression of sstr1, 3 and 5 was higher compared to the average in 23 other somatotropinomas (Table 1). Octreotide through receptor binding to sst2 moderately suppressed GH and IGF-1 levels in both patients. Switching to pasireotide improved hormonal control, suppressing GH and IGF-1 to the normal range, and even below the LLN. Pasireotide, which has higher affinity for sst5 and slightly lower affinity for sst2 than octreotide, was superior to octreotide in both patients. As both adenomas showed expression of sst2, the superiority of pasireotide over octreotide in hormonal suppression may be explained by an intracellular sub-receptor mechanism other than receptor abnormality.
Hyperglycaemia has previously been reported in patients receiving pasireotide,19 and following the switch to long-acting pasireotide both patients had some increase in glucose levels which were controlled with additional antidiabetic treatments in patient 1 and without medication in patient 2. Glucose homeostasis should be monitored carefully in patients receiving pasireotide and appropriate treatment initiated if glucose levels increase.20
In conclusion, following crossover from octreotide to pasireotide, our patients achieved long-term biochemical control, tumour volume reduction and improvement in the clinical signs and symptoms of acromegaly. Moreover, a reduction of pasireotide dose was possible due to IGF-1 over-suppression. Pasireotide-associated hyperglycaemia was appropriately managed in both patients with and without antidiabetic medications. IGF-1 over-suppression may be observed in a few patients and requires appropriate dose reduction of pasireotide; nevertheless, IGF-1 may still remain low.
COMPLIANCE WITH ETHICAL STANDARDS
Ethical approval
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the local institutional research committees and with the 1964 Helsinki declaration and its later amendments.
Acknowledgements
We thank Daniel Webber PhD of Mudskipper Business Ltd for medical editorial assistance with this manuscript.
FUNDING
The clinical studies were funded by Novartis Pharma AG. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation.
CONFLICT OF INTEREST
Ilan Shimon has received consulting, lectureship fees and research grants from Novartis, Chiasma and Pfizer. Monica R Gadelha has received consulting fees from Novartis and Ionis, lecture fees from Novartis and Ipsen and unrestricted research grants from Novartis, Ipsen and Pfizer. Wolfgang Saeger and Luiz Eduardo Wildemberg have nothing to declare.
REFERENCES
1. Melmed S 2006 Acromegaly. N Engl J Med 355: 2558-2573.2. Holdaway IM, Bolland MJ, Gamble GD, 2008 A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol 159: 89-95.
3. Giustina A, Chanson P, Bronstein MD, et al, Acromegaly Consensus Group 2010 A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab 95: 3141-3148.
4. Katznelson L, Atkinson JL, Cook DM, Ezzat SZ, Hamrahian AH, Miller KK, 2011 American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of acromegaly - 2011 update. Endocr Pract 17: Suppl 4: 1-44.
5. Schöfl C, Franz H, Grussendorf M, et al; Participants of the German Acromegaly Register, 2013 Long-term outcome in patients with acromegaly: analysis of 1344 patients from the German Acromegaly Register. Eur J Endocrinol 168: 39-47.
6. Chanson P, Borson-Chazot F, Kuhn J-M, Blumberg J, Maisonobe P, Delemer B, 2008 Control of IGF-I levels with titrated dosing of lanreotide Autogel over 48 weeks in patients with acromegaly. Clin Endocrinol (Oxf) 69: 299-305.
7. Melmed S, Cook D, Schopohl J, Goth MI, Lam KS, Marek J, 2010 Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel® therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension. Pituitary 13: 18-28.
8. Mercado M, Borges F, Bouterfa H, et al, SMS995B2401 Study Group, 2007 A prospective, multicentre study to investigate the efficacy, safety and tolerability of octreotide LAR® (long-acting repeatable octreotide) in the primary therapy of patients with acromegaly. Clin Endocrinol (Oxf) 66: 859-868
9. Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G, 2002 SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol 146: 707-716.
10. Colao A, Bronstein MD, Freda P, et al; Pasireotide C2305 Study Group, 2014 Pasireotide versus octreotide in acromegaly: a head-to-head superiority study. J Clin Endocrinol Metab 99: 791-799.
11. Gadelha M, Bronstein M, Brue T, et al; Pasireotide C2402 Study Group, 2014 Superior efficacy of pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly: randomized prospective Phase III study. Lancet Diabetes Endocrinol 2: 875-884.
12. Vieria Neto L, Wildemberg LE, Colli LM, et al, 2013 ZAC1 and SSTR2 are downregulated in non-functioning pituitary adenomas but not in somatotropinomas. PLoS One 8: e77406.
13. Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A, 2006 High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab 91: 4769-4775.
14. Colao A, Auriemma RS, Galdiero M, Lombardi G, Pivonello R, 2009 Effects of initial therapy for five years with somatostatin analogs for acromegaly on growth hormone and insulin-like growth factor-I levels, tumor shrinkage, and cardiovascular disease: a prospective study. J Clin Endocrinol Metab 94: 3746-3756.
15. Colao A, Auriemma RS, Pivonello R, Kasuki L, Gadelha MR, 2016 Interpreting biochemical control response rates with first-generation somatostatin analogues in acromegaly. Pituitary 19: 235-247.
16. van der Lely AJ, Biller BM, Brue T, et al, 2012 Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab 97: 1589-1597.
17. Bronstein MD, Fleseriu M, Neggers S, et al, Pasireotide C2305 Study Group, 2016 Switching patients with acromegaly from octreotide to pasireotide improves biochemical control: crossover extension to a randomized, double-blind, Phase III study. BMC Endocr Disord 16: 16.
18. Colao A, Bronstein MD, Brue T, et al, 2015 Pasireotide LAR maintains biochemical control in patients with cromegaly: results from the extension of randomised, phase III, PAOLA study. Endocr Abst 37: EP-810.
19. Petersenn S, Farrall AJ, Block C, et al, 2014 Long-term efficacy and safety of subcutaneous pasireotide in acromegaly: results from an open-ended, multicenter, Phase II extension study. Pituitary 17: 132-140.
20. Wildemberg LE, Gadelha MR, 2016 Pasireotide for the treatment of acromegaly. Expert Opin Pharmacother 17: 579-588.
Address for correspondence:
Ilan Shimon, MD, Institute of Endocrinology and Metabolism, Rabin Medical Center, Beilinson Hospital, Petah Tiqva 49100, Israel; Tel.: +972 54 574 9044, Fax: +972 3 937 7181, E-mail: ilanshi@clalit.org.il
Received: 31-01-2017, Accepted:22-02-2017