Department of Endocrinology, Diabetes and Metabolism, “Evaggelismos” Hospital, Athens, Greece
OBJECTIVE: To evaluate the therapeutic trends and long-term outcome of treatment modalities for acromegaly in our center over a 40-year period. Design: We retrospectively studied 321 acromegalic patients (145 males/176 females) diagnosed and treated from the 1970s until September 2013. Patients were divided into two subgroups: group A consisted of 166 patients diagnosed before 1990 and group B of 155 patients diagnosed after 1990. Outcome was assessed with GH (random and/or post OGTT) and IGF1 measurements. RESULTS: More group A than group B patients were submitted to radiotherapy (57.8% vs 16.8% patients, respectively, p <0.001). In contrast, more patients of group B were offered surgery (70.3% vs 42.1% in group A, p <0.001) and/or medical treatment (70.3% vs 23.4% in group A, p <0.001). At latest follow-up, 68.4 % of patients in group B achieved GH <2.5 μg/l after treatment vs 39.8% in group A, p=0.001, 46.9% of patients in group B achieved GH <1 μg/l vs 20.3% in group A, p=0.001 and 47.1% of patients in group B achieved during OGTT GH nadir <0.4 μg/l vs 18.6% in group A, p=0.001. CONCLUSIONS: Transsphenoidal resection and medical treatment resulted in improved outcome in acromegalic patients treated over the last 20 years. However, the disease still remains uncontrolled in a considerable number of patients.
Acromegaly, Medical treatment, Pituitary radiation, Somatostatin analogues, Surgery
INTRODUCTION
Acromegaly is a rare entity resulting from a GH secreting pituitary adenoma1 which, according to different multicentric registries, has a prevalence of around 60 cases per million2-4 and an incidence of 3 to 4 new cases per million annually,5 although a higher incidence of approximately 13 per 100,000 has also been reported.6 Increased morbidity and mortality result from the major complications i.e. cardiovascular disease, diabetes, hypertension and sleep apnea. Currently, surgery is the first-line treatment option.7 Medical therapy, previously applied mainly as an adjuvant treatment modality, is nowadays increasingly used as primary therapy in selected patients with macroadenomas with low probability of surgical remission.7 The use of pituitary irradiation, once widely applied, is currently limited to a few cases.7 Decisions regarding therapy should take into account costs, availability and expertise of the abovementioned therapeutic options. Given the rarity of the disease, epidemiological data regarding treatment modalities and long-term outcome through national acromegaly registries are valuable in order to improve our understanding of existing interventions in relation to clinical outcome. Such a registry has been in existence since the early 1970s in our department, a major pituitary referral center in Greece. The aim of the present study was to examine the therapeutic trends and long-term outcome of acromegaly in our center over a 40-year period. We also aimed to evaluate the effect of somatostatin analogues on tumor size reduction and compressive phenomena and to identify possible risk factors that affect their antitumor activity.
PATIENTS AND METHODS
Study population
We retrospectively studied 321 acromegalic patients (145 males/176 females) diagnosed and treated in our Department from 1970 until September 2013. The mean time of follow-up was 94.9±107.4 months (from 12 to 549, median 68 months). Only patients who had been followed up for at least 12 months after diagnosis were included. Exclusion criteria were pregnancy, presence of chronic kidney, liver or psychiatric disease and ectopic GHRH production; in the event of pregnancy or of the above chronic diseases the interpretation of GH and IGF1 results would be unreliable. Hospital charts were reviewed by three physicians who also performed data entry into a computerized database platform developed using Access 97 software (Microsoft Corporation, 1996) and kindly donated by Dr S. Melmed.8
Diagnosis was based upon clinical picture and biochemical confirmation including non-suppressed GH after the oral glucose tolerance test (OGTT) and/or elevated IGF-1. Imaging techniques were computed tomography (CT) until 1990 and magnetic resonance imaging (MRI) afterwards. In patients diagnosed before CT implementation in our center (1980), diagnosis of macroadenoma was based upon radiological estimation of the sella, while in the presence of clinical and biochemical evidence of acromegaly with normal sella, the diagnosis of microadenoma was assigned.
Treatment modalities included surgery via transsphenoidal and in a very few cases via the transfrontal approach, pituitary irradiation (conventionally by means of a linear accelerator, or more recently by stereotactic radiosurgery, namely linear-accelerator based Cyber-knife or cobalt-60 radiation source Gamma-knife) and medical therapy i.e. dopamine agonists (Parlodel, Meda Pharmaceuticals, Bishop’s Stortford, UK and Dostinex, Pfizer, Kent, UK) somatostatin analogues (Sandostatin® LAR®, Novartis Pharmaceuticals, Surrey, UK and Somatulin® Autogel®, Ipsen, Berkshire, UK) or pegvisomant (Somavert, Pfizer, Kent, UK). Outcome was evaluated by GH measurements: GH <2.5 μg/l, or GH <1 μg/l (random measurement or mean value of at least two basal samples) or GH post OGTT <0.4 μg/l. After 1990, when IGF1 measurement became widely available in our department, normal for age IGF1 was added to the outcome criteria.7-9 Overall disease control was defined as GH <2.5 μg/l and normal IGF1. Thyroid, adrenal and gonadal functions were appropriately assessed and substituted when deficient.
Patients were divided into two subgroups according to the time of diagnosis: group A consisted of 166 patients diagnosed before 1990, while group B consisted of 155 patients diagnosed after 1990. This threshold was set in order to evaluate the impact of modern imaging techniques, i.e. the introduction of MRI at this time point in the diagnosis and treatment of acromegalic patients. Additional analysis was performed for each decade of enrollment. In a subgroup of 55 acromegalic group B patients (aged 52.1±1.6 years, 18 males and 37 females) antitumor activity of somatostatin analogues was evaluated; these patients were treated with somatostatin analogues before or after unsuccessful surgery and did not receive irradiation. As all patients in this subgroup were diagnosed after 1990, tumor size was estimated by magnetic resonance imaging (MRI). Tumor shrinkage was defined as a 20% decrease in the largest measurable adenoma diameter.
Hormonal assays
Samples were collected and centrifuged and serum was stored in aliquots at -200C until assayed. GH and IGF1 assays used in our department have changed over the years. Serum GH levels were measured by RIA until 1995 and thereafter by IRMA assay kits (Pharmacia, Uppsala, Sweden until 2000 and CIS Bio International, Gif-sur-Yvette, France, thereafter). The CIS assay standards are calibrated against the 2nd International Standard WHO IS 98/574. This assay has a detection limit of 0.04μg/l, and the intra-assay coefficient of variation is 2.4% and 2.8% at a concentration of 3.5μg/l and 17μg/l, respectively. Serum IGF-1 was measured with RIA from 1990-1998 and with IRMA thereafter (1998-2002 by Nichols Institute, San Juan Capistrano, CA, USA and since 2002 by Mediagnost, Reutlingen, Germany). The standards of the Mediagnost assay are calibrated against the International Standard WHO NIBSC 02/254. To dissociate IBNIGF-I from the binding proteins, samples were diluted in an acidic buffer and then exposed to antiserum containing an excess of IGF-II to block the binding proteins. The detection limit of the assay is 0.1μg/l. The intra-assay coefficient of variation is 2.5-3.4% and the inter-assay coefficient of variation is 4.5-6.2%. Thyroid, adrenal and gonadal functions were regularly assessed by evaluating basal thyroxine (T4) or free T4, cortisol, luteinising hormone (LH), follicle-stimulating hormone (FSH), testosterone or estradiol.
Statistical analysis
Variables were expressed throughout as mean±SD. Statistical analysis was performed using the SigmaStat statistical package (version 4.0, 2008 Systat Software, San Jose, CA.) The one-sample Kolmogorov-Smirnov test was used to test whether a variable was normally distributed. Statistical analysis to identify differences between the two groups was performed by t-test, or the Mann-Whitney rank sum test when data distribution was not normal. Univariate and multivariate were performed to identify predictors of tumor response to somatostatin analogues. The chi square (χ2) test was performed to compare non-parametric variables and the Z-test to compare proportions. Spearman correlations were used to examine the relation variables. The level of significance was set at 0.05 for all statistical tests.
RESULTS
Demographic, clinical and biochemical data of patients
As depicted in table 1, 45.2% of our patients were men and 54.8% women. The mean age at diagnosis was 45.5±12.5 years. Microadenomas were diagnosed in 158 (49.2%) and macroadenomas in 163 (50.8%) patients. The delay of diagnosis (time from onset of symptoms as depicted by patients’ history or photos until diagnosis) was 85.7±64.5 months. In group B more macroadenomas were identified compared to group A (61.7 vs 40.4, p=0.001). Time of follow-up was 139.5±144.4 months in group A and 62.1±61.3 (p=0.001) in group B. No statistically significant differences were found in age at diagnosis, sex and delay of diagnosis between groups A and B, although group B patients were almost 6 years older than group A patients. Most patients were referred to our department by endocrinologists (63.5%) and internists (17.1%). As revealed by distribution comparisons, the prevalence of acromegaly was higher between 30 and 60 years of age: 233 patients (69.8%) vs 46 patients (14.3%) <30 years and 42 patients (13.1%) >60 years, p <0.001.
Treatment modalities
As shown in Figure 1, there was a significant change in the treatment modalities offered to our patients during the observation period. More group A than group B patients were submitted to radiotherapy (57.8% vs 16.8% patients, respectively, p <0.001); in contrast, more patients of group B were offered surgery (70.3% vs 42.1 group A, p <0.001) and/or medical treatment (70.3% vs 23.4% group A, p <0.001).
The analysis of treatment modalities data per the four time periods (1970-1980, 1981-1990, 1991-2000, 2001-2013) is shown in Figure 1. The use of pituitary irradiation was significantly reduced in the more recent periods (offered to 61.2%, 33.6%, 16.8% and 4.3% of patients for the 1st, 2nd, 3rd and 4th periods, respectively, p <0.001 for all comparisons). Surgery was offered to statistically more patients in the 2nd and 3rd compared to the 1st period (43.9% and 52.8% vs 23.5% of patients, respectively, p <0.001 for both comparisons). Finally, the use of medical treatment increased significantly during the 4th period (58.9% vs 15.3%, 22.4%, 30.4% of patients during the 1st, 2nd and 3rd period, respectively, p <0.001 for all comparisons).
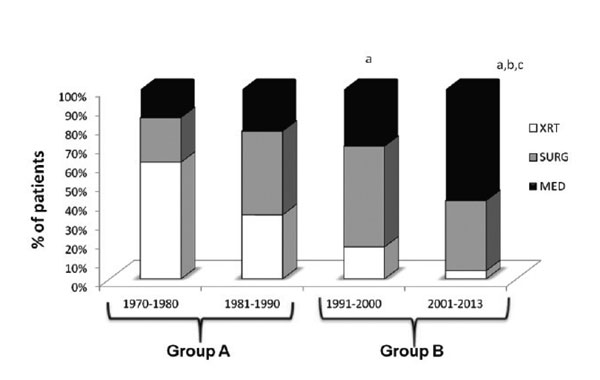
Figure 1. Evolution of treatment modalities of acromegaly during 4 time periods a: denotes statistically significant difference from 1st time period b: denotes statistically significant difference from 2nd time period c: denotes statistically significant difference from 3rd time period p <0.05.
More specifically, surgical treatment was offered to a total of 179 patients (170 with the transsphenoidal and 9 with the transfrontal approach); in 137 patients (76.5%) surgery was used as first-line treatment (58 in group A and 79 in group B), in 38 patients (21.2%) post-medical treatment (8 in group A and 30 in group B) and in 4 (2%, all in group A) after pituitary radiation.
Radiation therapy was offered to 122 patients, 117 conventional (who received 3500-6000 cGy) and 5 stereotactic radiosurgery (who received 2500-5000 cGy), mean irradiation dose 4560±517 cGy; as first-line treatment in 82 patients (67.2%, 76 in group A and 6 in group B) and as second-line treatment in 40 patients (32.7%, 14 patients in group A and 26 in group B).
Medical treatment was offered to 146 patients: in 84 patients (57.5%) as first-line approach (9 in group A and 75 in group B patients) and in 62 patients (42.4%, 30 group A and 32 in group B) after surgery or radiation failure. Somatostatin analogues were used in 113 patients, while 53 patients received dopamine agonists (either cabergoline or bromocriptine, 33 as monotherapy and 20 in combination with somatostatin analogues). More specifically, among the 113 patients administered somatostatin analogues, 34% received lanreotide: 11.7% at the dose of 60 mg, 7.7% at the dose of 90 mg, 14.6% at the dose of 120 mg every 4 weeks. The remaining 66% were administered octreotide LAR: 24.3% at the dose of 20 mg and 41.7% at the dose of 30 mg every 4 weeks. Regarding dopamine agonists, 40 patients received bromocriptine (mean daily dose 12 mg) and 13 patients cabergoline (mean weekly dose 2.4 mg. Finally, 13 patients received pegvisomant (mean daily dose 16.5 mg), 4 as combined treatment with somatostatin analogues and 9 as monotherapy after failure of somatostatin analogues to control the disease.
Acromegaly outcome
The final outcome of acromegaly in our patients is presented in Figure 2. Data regarding treatment modalities applied were available in 299 out of 321 patients (the remaining 22 were either not yet treated or lost to follow-up before any therapeutic intervention). As demonstrated in Figure 2, in 119 (37%) of our patients various treatment combinations were attempted. Overall, 55.7% of patients achieved GH <2.5μg/l, 35.1% of patients achieved GH <1μg/l, 34% achieved GH <0.4 μg/l and 70.3% normalized IGF1. When comparing the effectiveness of the various treatments used, a similar outcome was found between patients submitted to surgical or medical treatment as first-line approach or combination of surgical, medical and/or irradiation treatment after unsuccessful first-line approach (Figure 2). Notably, all treatment groups were similar in terms of tumor size.
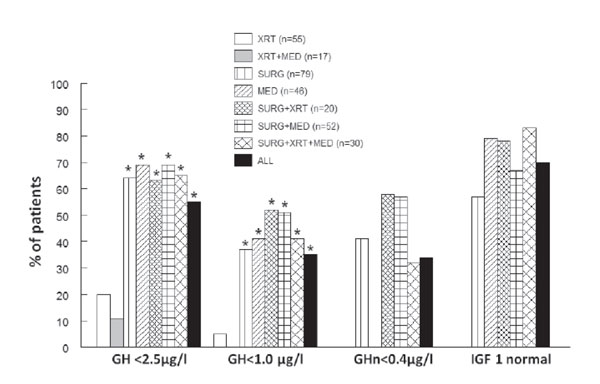
Figure 2. Disease outcome with different treatment modalities in our cohort of acromegalic patients (*) denotes statistically significant difference from patients who received radiation (XRT) or radiation+medical treatment (XRT+MED).
Irradiation as monotherapy or followed by medical treatment led to worse outcome compared to all other treatment groups (GH <2.5 μg/l in 20% and 11.7% of respectively, p <0.001 for all comparisons). During the observation period lasting 11.42 years (range 2.4-43.4 years) no report of radionecrosis, cerebrovascular disease or second tumor formation was noted in those patients submitted to irradiation. At the end of the observation period pituitary function tests following irradiation were available in 49 patients; when compared to 132 non-irradiated patients a higher incidence of pituitary deficiencies was noted: FSH/LH deficiency was found in 63.3% irradiated vs 12.4% non-irradiated patients (p <0.001), ACTH deficiency in 55.5% vs 7.5% (p <0.001) and TSH deficiency in 39.9% vs 6.8% (p <0.001).
When dopamine agonists were used as monotherapy control rate was low: 30.4% achieved GH less than 2.5μg/l and only 13% GH less than 1μg/l (IGF1 data are few and not shown). When combined with somatostatin analogues the control rate was higher: 57.8% achieved GH <2.5μg/l, 21% GH <1μg/l and 64.7% normal IGF1. Somatostatin analogues therapy resulted in 76.3% of patients achieving GH <2.5μg/l, 50% achieving GH <1μg/l and 71.8% normalizing IGF1. Furthermore, 75% of patients on pegvisomant either as monotherapy or in combination with somatostatin analogues normalized IGF1.
Analysis of disease outcome per time of diagnosis is shown in Figure 3: 68.4% of patients in group B achieved GH <2.5μg/l after treatment vs 39.8% in group A, p=0.001, 46.9% of patients in group B achieved GH <1μg/l after treatment vs 20.3% in group A, p=0.001 and 47.1% of patients in group B achieved during OGTT GH nadir <0.4μg/l after treatment vs 18.6% in group A, p=0.001. IGF1 data were available in 38 group A patients vs 127 group B patients, thus IGF1 data for group A patients are not shown; 60.7% of group B patients achieved disease control (GH <2.5μg/l and normal IGF1).
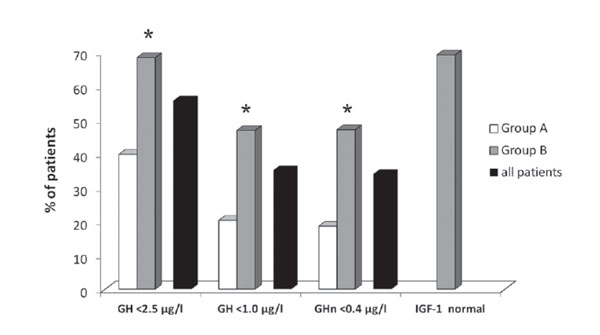
Figure 3. Overall disease outcome in the two time groups of acromegalic parients (*): denotes statistically significant difference from group A, p <0.05 GH <2.5, GH <0.1: random GH levels <2.5µg/L and <1.0µg/L respectively, GH <0.4: GH nadir levels in the glucose suppression test <0.4 µg/L.
A subgroup of 55 patients was treated with
somatostatin analogues and did not receive radiation therapy. As shown in Table
2, 24 patients (43.6%) demonstrated shrinkage of the pituitary adenoma by 33.3%
in response to therapy. Responders had statistically higher prevalence of
macroadenomas (83.3% vs 51.6%, p=0.030), larger adenoma size (17.1±9.8 vs
10.5±7.8mm p <0.001) and higher pre-treatment GH levels (20.2±16.6μg/l vs
13.2±11.9μg/l p=0.043) compared to non-responders (Table 2). Furthermore, 70.8%
of the responders achieved biochemical control vs 25.8% of the non-responders,
p <0.001). In 40 patients somatostatin analogues were given as first-line
treatment and in 15 as second-line treatment after transsphenoidal resection.
Somatostatin analogues were equally effective in inducing tumor size reduction
in both groups (47.5% in the first-line treatment group and 33.3% in the
second-line treatment group). Tumor shrinkage was positively correlated to
pre-treatment size in both groups (r=0.47, p=0.024 for first-line treatment and
r=0.63, p=0.011 for second-line treatment) as depicted in Figure 4. 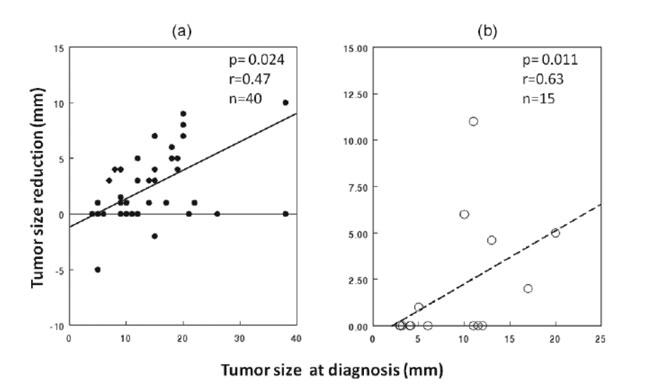
Figure 4. Correlation of tumor size reduction with initial tumor size in patients to whom somatostatin analogues were given as (a) first-line treatment (b) second-line treatment after transsphenoidal resection.
In order to identify predictors of tumor response to somatostatin analogues we performed univariate logistic regression analyses. The following factors were able to predict response: pretreatment adenoma size (odds ratio 1.11, 95%CI: 1.01-1.21, p=0.018) and biochemical control of acromegaly under treatment; patients with biochemical control had an odds ratio of having tumor shrinkage of 7.0 (95%CI: 2.1-23, p=0.001). Multivariate logistic regression analysis showed that both factors were independent predictors of tumor response to somatostatin analogues (pretreatment tumor size; odds ratio 1.15, 95%CI: 1.03-1.28, p=0.013, control of acromegaly odds ratio 9.75, 95%CI: 2.40-39.47, p=0.001). Pretreatment GH levels were related to tumor size (r=0.36, p=0.011) and were not independently associated with response of the tumor to SSAs. Before somatostatin treatment, 4 patients had visual field defects; 3 of them experienced tumor size reduction under treatment and 2 restored visual fields, while in one patient both tumor size and visual fields remained unchanged.
Finally, the outcome of 32 acromegalic patients with prolactin co-secretion was evaluated. After surgical treatment (offered to 28 patients), 70% of patients achieved GH <2.5μg/l, 40% achieved GH <1μg/l, 40% achieved GH <0.4μg/l and 55% normalized IGF1. Medical treatment (cabergoline and somatostatin analogues) was offered to all patients with uncontrolled disease after surgery as well as to the 4 patients who did not have surgery. Overall, 75% of patients achieved GH <2.5μg/l, 45% of patients achieved GH <1 μg/l, 45% achieved GH nadir <0.4μg/l during OGTT and 60% normalized their IGF1.
DISCUSSION
In this study a survey of the treatment and outcome of acromegaly in our department is presented. Age distribution, sex and age of diagnosis are similar to other published databases.10-16 Delay of diagnosis was approximately 7 years, as also observed in the French and Italian acromegaly registries,3,15 and according to our findings no improvement was noted over the last 20 years. Micro- and macroadenoma distribution in our cohort was almost equal in contrast to other surveys where macroadenomas account for 67.4% to 83% of the total.3,10,12,13 The higher prevalence of microadenomas recorded especially in patients evaluated before 1990 probably reflects the lack of sensitive imaging techniques leading to underestimation of tumor dimensions.
A clear finding of our study is the change in therapeutic modalities over the observation period, from the common use of irradiation before 1980 to the gradually increasing use of surgery and medical treatment with a parallel decline in the use of irradiation thereafter. A similar pattern of therapy trends has been reported by others.11,12,17 Of note, surgery was offered less commonly to our patients (55.7%) compared to other studies reporting 68% to 91% surgery in their cohorts,10-14,18 possibly reflecting a relative lack of pituitary surgery centers in our country. The incidence of pituitary irradiation in our center was comparable to those reported elsewhere.10,17 Similarly, the use of medical or combined treatments in our cohort is in agreement with published data from other centers.10-13,15,16,18
In the long-term follow-up after surgical treatment, 64.5% of our patients achieved GH <2.5μg/l, 37.5% GH <1μg/l, while 57.1% of surgically treated patients normalized IGF1 levels. Schoelf et al13 reported a surgical cure rate of 38.8% using only the IGF1 criterion. In other large cohorts from Belgium and Spain, surgical success rate is 46.5% and 34%, respectively, according to both GH <2μg/l and IGF1 criteria.12,16 A possible explanation for this discrepancy is the high percentage of microadenomas in our registry, in which transsphenoidal surgery is often curative.19
According to our findings, irradiation either alone or followed by medical treatment fails to achieve safe GH levels, i.e. <2.5μg/l (20% and 11.7%, respectively). We have previously shown that half of the patients achieve GH <5μg/l even after 10 years following radiation therapy and that safe GH levels of less than 1 μg/l are infrequently achieved.20 Higher response rates from other studies,12,13,21 ranging from 34.8-77%, are due to selection bias, since in the vast majority of these subjects radiation therapy was used as second-line treatment after surgery failure.
Primary medical treatment resulted in GH <2.5 μg/l in 69.4% of patients, GH <1μg/l in 41.6% and normal IGF1 in 79.4%. This control rate is comparable to that reported by Schoefl et al (65.2% with normal IGF1)13 and higher than that reported by Bex et al (24% GH <2μg/l and normal IGF1),12 which according to the authors might be due to lower patient compliance and differences in somatostatin analogues dosing. A recent meta-analysis including treatment naïve patients with acromegaly reports remission rates up to 45% using at least one biochemical remission criterion, i.e. either GH <2.5μg/l or normal IGF1.22
The efficacy of somatostatin analogues used either as primary therapy or in combined treatment modalities is in good agreement with other published series23-26 (44-72% for GH levels <2.5μg/l and 38-75% for normal IGF1). By contrast, the relatively low control rate (75%) under pegvisomant in this study, compared to the optimal 97% reported by Neggers et al,27 probably reflects the low mean daily dose (16.5mg)28 or possibly lack of compliance.
We have shown that disease outcome has improved over the last 20 years, reflecting the change in therapeutic trends in favor of surgery and medical treatment. Indeed, in agreement with other studies,11,12 multimodality treatment is often necessary and guarantees the best possible outcome (65.5% GH <2.5μg/l, 41.3% GH <1μg/l, 31.8% GH <0.4μg/l and 82.6% normal IGF1) (Figure 2).
It has been reported that administration of somatostatin analogues may result in tumor shrinkage, especially if given as first-line treatment, i.e. before pituitary surgery;29,30 however, their antisecretory and antiproliferative effect may be dissociated.31 In 43.6% of our patients somatostatin analogues (as primary or as adjuvant therapy) reduced tumor size by 33.3%, an outcome compatible with a 53% respond rate in a recent meta-analysis.32 A positive correlation between tumor shrinkage and biochemical response was revealed, also noted in our study. In contrast, Caron et al33 in a prospective study using somatostatin analogues as primary therapy have shown a higher response rate (62.9%), a difference that could be explained by the difference between study design (retrospective vs prospective). Finally, in our population the degree of tumor shrinkage depended on initial tumor size as already shown by others.26,34,35
The strength of this study representing real-life experience in the management of acromegaly lies in the single center experience and thus the same treatment practices in each time period, although not all patients were operated on in the same neurosurgical unit. A major limitation is the use of different assays for GH and IGF1 measurements over 4 decades and the lack of data regarding mortality.
In summary, our analysis illustrates the theurapeutic trend in favor of transsphenoidal resection and somatostatin analogues and reduction of use of radiation over the years. This change has led to improved outcome. However, in a substantial proportion of patients, disease still remains uncontrolled demanding close follow-up and the use of multimodality treatment.
CONFLICT OF INTEREST
S. Tsagarakis received honoraria, travel grants and participated in advisory boards of Novartis, Ipsen and Pfizer. M. Tzanela received travel grants from Novartis, Ipsen and Pfizer.
REFERENCES
1. Melmed S, 2009 Acromegaly: pathogenesis and treatment. J Clin Invest 119: 3189-3202.2. Sesmilo G, 2013 Epidemiology of acromegaly in Spain. Endocrinol Nutr 60: 470-474.
3. Fieffe S, Morange I, Petrossians P, et al, 2011 Diabetes in acromegaly, prevalence, risk factors and evolution: data from the French acromegaly registry. Eur J Endocrinol 164: 877-884.
4. Petrossians P, Tichomirowa MA, Stevenaert A, Martion D, Daly AF, Beckers A, 2012 The liege acromegaly survey (LAS); a new software tool for the study of acromegaly. Ann Endocrinol Paris 73: 193-201.
5. Holdaway IM, Rajassorya C, 1999 Epidemiology of acromegaly. Pituitary 2: 29-41.
6. Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A, 2006 High prevalence of pituitary adenomas: a cross-sectional study in the provence of Liege, Belgium. J Clin Endocrinol Metab 91: 4769-4775.
7. Katznelson L, Raws ERJr, Melmed S, et al, 2014 Acromegaly: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 99: 3933-3951.
8. Drange MR, Fram MR, Herman-Bonert V, Melmed S, 2000 Pituitary tumor registry: A novel clinical resource. J Clin Endocrinol Metab 85: 168-174.
9. Holdaway IM, Rajassorya RC, Gamble GD, 2004 Factors influencing mortality in acromegaly. J Clin Endocrinol Metab 89: 667-674.
10. Kauppinen-Mäkelin R, Sane T, Reunanen A, et al, 2005 A nationwide survey of mortality in acromegaly. J Clin Endocrinol Metab 90: 4081-4086.
11. Vallette S, Ezzat S, Chik C, et al, 2013 Emerging trends in the diagnosis and treatment of acromegaly in Canada. Clin Endocrinol (Oxf) 79: 79-85.
12. Bex M, Abs R, T’Sjoen G, et al, 2007 AcroBel – the Belgian registry on acromegaly: a survey of the ‘real-life’ outcome in 418 acromegalic subjects. Eur J Endocrinol 157: 399-409.
13. Schofl C, Franz H, Grussendorf M, et al, 2012 Long-term outcome in patients with acromegaly: analysis of 1344 patients from the German Acromegaly Register. Eur J Endocrinol 168: 39-47.
14. Kwon O, Song YD, Kim SY, et al, 2013 Nationwide survey of acromegaly in South Korea. Clin Endocrinol (Oxf) 78: 577-585.
15. Arosio M, Reimondo G, Malchiodi E, et al, 2012 Predictors of morbidity and mortality in acromegaly: an Italian survey. Eur J Endocrinol 167: 189-198.
16. Mestron A, Webb SM, Astorga R, et al, 2004 Epidemiology, clinical characteristics, outcome, morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry. Eur J Endocrinol 151: 439-446.
17. Sesmilo G, Gaztambide S, Venegas E, et al, 2013 Changes in acromegaly treatment over four decades in Spain: analysis of the Spanish Acromegaly Registry (REA). Pituitary 16: 115-121.
18. Cuevas-Ramos D, Carmichael JD, Cooper O, et al, 2015 A structural and functional acromegaly classification. J Clin Endocrinol Metab 100: 122-131.
19. Jane Jr, JA, Starke RM, Elzoghby MA, et al, 2011 Endoscopic Transsphenoidal Surgery for Acromegaly: Remission Using Modern Criteria, Complications, and Predictors of Outcome. J Clin Endocrinol Metab 96: 1-9.
20. Thalassinos NC, Tsagarakis S, Ioannides G, et al, 1998 Megavoltage pituitary irradiation lowers but seldom leads to safe GH levels in acromegaly: a long-term follow-up study. Eur J Endocrino 138: 160-163.
21. Jenkins PJ, Bates P, Carson MN, et al, 2006 Conventional pituitary irradiation is effective in lowering serum growth hormone and insulin-like growth factor-I in patients with acromegaly. J Clin Endocrinol Metab 91: 1239-1245.
22. Abu Dabrh AM, Mohammed K, Asi N, et al, 2014 Surgical Interventions and Medical Treatments in Treatment-Naïve Patients With Acromegaly: Systematic Review and Meta-Analysis. J Clin Endocrinol Metab 99: 4003-4014.
23. Mercado M, Borges F, Bouterfa H, et al, 2007 A prospective, multicentre study to investigate the efficacy, safety and tolerability of octreotide LAR (long-acting repeatable octreotide) in the primary therapy of patients with acromegaly. Clin Endocrinol (Oxf) 66: 659-678.
24. Carmichael J, Bonert VS, Nuno M, et al, 2014 Clinical Trial Methodology Impact on Reported Biochemical Efficacy Rates of Somatostatin Receptor Ligand Treatments: A Meta-Analysis. J Clin Endocrinol Metab 99: 1825-1833.
25. Cozzi R, Montini M, Attanasio R, et al, 2006 Primary treatment of acromegaly with octreotide LAR: a long-term (up to nine years) prospective study of its efficacy in the control of disease activity and tumor shrinkage. J Clin Endocrinol Metab 91: 1397-1403.
26. Cozzi R, Attanasio R, Montini M, et al, 2003 Four-Year Treatment with Octreotide-Long-Acting Repeatable in 110 Acromegalic Patients: Predictive Value of Short-Term Results by ROC analysis. J Clin Endocrinol Metab 88: 3090-3098.
27. Neggers SJ, Franck SE, de Rooij FW, et al, 2014 Long-term efficacy and safety of pegvisomant in combination with long-acting somatostatin analogs in acromegaly. J Clin Endocrinol Metab 99: 3644-3652.
28. Higham CE, Chung TT, Lawrance J, et al, 2009 Long-term experience of pegvisomant therapy as a treatment for acromegaly. Clin Endocrinol (Oxf) 71: 86-91.
29. Freda PU, Katznelson L, van der Lely AJ, et al, 2005 Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab 90: 4465-4473.
30. Bevan JS, 2005 Clinical review: the antitumoral effects of somatostatin analog therapy in acromegaly. J Clin Endocrinol Metab 90: 1856-1863.
31. Resmini E, Dadati P, Ravetti JL, et al, 2007 Rapid pituitary tumor shrinkage with dissociation between antiproliferative and antisecretory effects of a long-acting octreotide in an acromegalic patient. J Clin Endocrinol Metab 92: 1592-1599.
32. Giustina A, Mazziotti G, Torri V, et al, 2012 Meta-analysis on the effects of octreotide on tumor mass in acromegaly. PLoS One 7: e36411.
33. Caron PJ, Bevan JS, Petersenn S, et al, 2014 Tumor Shrinkage With Lanreotide Autogel 120 mg as Primary Therapy in Acromegaly: Results of a Prospective Multicenter Clinical Trial. J Clin Endocrinol Metab 99: 1282-1290.
34. Amato G, Mazziotti G, Rotondi M, et al, 2002 Long-term effects of lanreotide SR and octreotide LAR on tumour shrinkage and GH hypersecretion in patients with previously untreated acromegaly. Clin Endocrinol (Oxf) 56: 65-71.
35. Plöckinger U, Reichel M, Fett U, Saeger W, Quabbe HJ, 1994 Preoperative octreotide treatment of growth hormone-secreting and clinically nonfunctioning pituitary macroadenomas: effect on tumor volume and lack of correlation with immunohistochemistry and somatostatin receptor scintigraphy. J Clin Endocrinol Metab 79: 1416-1423.
Address for correspondence:
S. Tsagarakis MD, PhD, FRCP, 45-47 Ipsilantou Street, 10676 Athens, Greece; Fax: +30 213 2041828, E-mail: stsagara@otenet.gr
Received:16-02-2016, Accepted: 31-05-2016