1Division of Endocrinology and Diabetology, Department of Internal Medicine II, University Hospital of Freiburg, Freiburg, Germany; 2Department of Internal Medicine I, St. Vincentius-Hospital, Karlsruhe, Germany; 3Department of Pathology, Institute of Surgical Pathology, University Hospital of Freiburg, Freiburg, Germany; 4Division of Endocrine Surgery, Department of General and Visceral Surgery, University Hospital of Freiburg, Germany; 5School of Biosciences, Cardiff University, Cardiff, United Kingdom; 6Division of Endocrine Surgery Hospital Reinbek St. Adolf-Stift Reinbek, Germany; 7Endocrine Practice and Molecular Laboratory, Heidelberg, Germany
BACKGROUND:
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal-dominant hereditary
disorder associated with the development of endocrine tumors due to reduced
expression of the tumor suppressor protein menin. Recent studies indicate a
general role of menin in carcinogenesis, affecting the prevalence and clinical
course of common non-endocrine tumors such as breast cancer, hepatocellular
carcinoma and melanoma. Here we report a new germline missense mutation of Men1
in a German family with atypical tumor phenotype over three generations. Based
on the type of mutation, we discuss possible changes in menin function leading
to atypical tumorigenesis and present the clinical significance of such
findings.
CASE PRESENTATION: A German family with a history of primary hyperparathyroidism
presented to our Hospital for further evaluation. Members of the family
demonstrated many different atypical tumors, such as renal cell carcinoma,
papillary thyroid cancer and prostate cancer. DNA sequencing from peripheral
blood revealed a novel mutation: Ser38Cys [TCC>TGC] in exon 2, codon 38 of Men1.
This novel mutation is located in a region of menin which is responsible for
interactions with the transcription factor JunD. This factor has recently been
associated with prostate cancer. DNA sequencing of two of the atypical tumors
(prostate cancer, papillary thyroid cancer) did not reveal a loss of
heterozygosity, indicating an impact on menin expression and function in the
heterozygous state, in line with results in +/-Men1 mutant
mice developing prostate cancer.
CONCLUSION: The results and clinical course of
disease in this case indicate the potential role of menin in the development of
non-endocrine or atypical-endocrine tumors in MEN1 patients. Further
investigations are needed to clarify both the general role of menin and the
importance of specific mutations in carcinogenesis. Nevertheless, in families
with uncommon manifestations of the syndrome early diagnostic adjustments
should be considered.
Atypical tumors, MEN1, New mutation, Prostate cancer
INTRODUCTION
Multiple endocrine neoplasia type 1 (MEN1) is a rare autosomal-dominant hereditary endocrine tumor syndrome associated with reduced levels of the tumor suppressor protein menin. Results from clinical studies and preclinical experiments indicate an additional involvement of Men1, which encodes menin, in the development of non-endocrine tumors.1 Herein we report a novel mutation of Men1 followed over three generations of a German family with atypical non-endocrine tumor entities.
CASE DESCRIPTION
A 75-year-old man (patient 1) presented to our hospital with his 47-year-old son (patient 2) with primary hyperparathyroidism (PHPT). Patient 1 had been previously diagnosed with: a) papillary thyroid cancer with multiple thyroid nodules at the age of 72, treated with total thyroidectomy and radiation, b) renal cell carcinoma at the age of 72, treated with partial kidney resection, c) prostate cancer at the age of 62, treated with prostatectomy and d) PHPT at the age of 72, treated with total parathyroidectomy. At the first visit we detected elevated chromogranin A levels. Gastroscopy, colonoscopy, repeated DOTATATE-PET-CTs and annual abdominal MRIs did not locate a gastrointestinal tumor. An MRI of the pituitary gland at the fifth annual follow-up revealed a microadenoma, classified as a non-secreting tumor by inconspicuous hormonal results. Additionally to PHPT diagnosed at the age of 44, patient 2 was diagnosed with: a) cold thyroid nodule at the age of 35, treated with total thyroidectomy, and b) ulcerative colitis at the age of 42. PHPT was treated initially with partial parathyroidectomy and on relapse with total parathyroidectomy and autotransplantation of one parathyroid gland to the brachioradialis muscle. Furthering the family history, the brother of patient 2 (patient 3) and daughter of patient 3 (patient 4) were both diagnosed at the first screening for MEN1 syndrome with PHPT at the ages of 46 and 21, respectively, and subsequently operated on. Interestingly, patient 3 was also diagnosed with prostate cancer at the age of 46 and was treated with prostatectomy (Figure 1). Patients 3 and 4 had follow-up visits in another clinic.
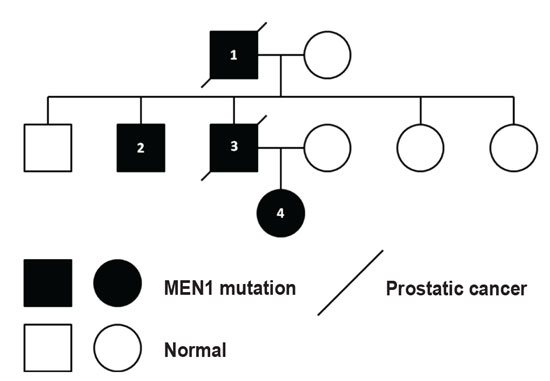
Figure 1. Patients 1-4 developed a PHPT and patients 1 and 3 cold thyroid nodules. Patients 1 and 3 additionally developed prostate cancer early in life. Patient 1 was also diagnosed with a papillary thyroid cancer and a renal cell carcinoma.
The genetic diagnosis of peripheral blood from patients 1 and 2 revealed a novel heterozygous missense mutation: Ser38Cys (TCC>TGC) in exon 2, codon 38 of Men1 (Figure 2). Additional analysis by DNA sequencing and multiplex ligation-dependent probe amplification from the papillary thyroid carcinoma of patient 1 and from the prostate cancer of patient 3 did not demonstrate a “Loss of Heterozygosity” (LOH).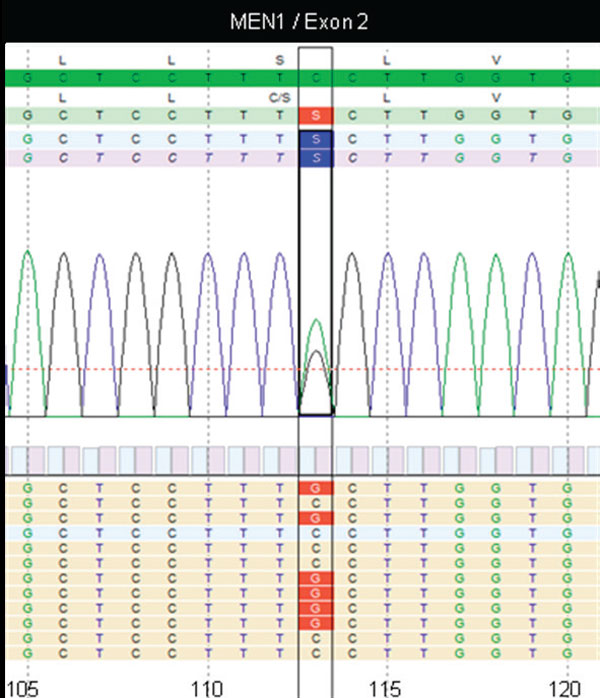
Figure 2. A novel missense mutation in Men1/Exon 2 c.113>G, p.Ser38Cys associated with endocrine and very probably atypical non-endocrine tumor entities over three generations of a German family.
DISCUSSIONThe management of families with MEN1 syndrome is often very demanding. The clinical guidelines concentrate on the early detection of the three most common tumor manifestations, namely parathyroid adenoma, enteropancreatic neuroendocrine tumors and anterior pituitary adenomas. Other features are either rare (e.g. adrenal cortical tumors) or clinically less important (e.g. lipomas, angiofibromas).1,2 However, different atypical tumor entities have occasionally been described in families with MEN1 syndrome,3-6 raising the following questions. Does menin have a major role in carcinogenesis, including a non-endocrine function? Do some MEN1-families with specific mutations have an increased risk of non-endocrine tissue malignancies, which may demand diagnostic adjustments?
In vivo, -/-Men1 mice die embryonically, while +/-Men1 mice develop mostly endocrine tumor disorders, such as parathyroid hyperplasia, and pancreatic, pituitary and adrenal cortical tumor.7 Menin is however expressed not only in endocrine tumors but also in many non-endocrine human cancer cells, such as in breast, cervical, brain glioma and gastric cancer cell lines.8 Furthermore, changes in menin expression have recently been related to the development of non-endocrine tumors. A downregulation of menin is observed in osteosarcoma tissues.9 Additionally, MEN1 patients more frequently develop malignant melanomas. Menin suppresses the expression of the growth factors pleiotrophin and phosphatidylinositol 3-kinase, inhibiting melanoma cell proliferation and migration. Moreover, menin stimulates the transcription of genes involved in DNA repair.5,10 By contrast, increased menin levels promote hepatocellular carcinogenesis correlating negatively with the overall and tumor-free survival rates.6 In our case, two of the three male carriers of the Men1 mutation developed prostate cancer early. Sequencing of prostate tumor cells from patient 3 excluded LOH, which could explain these early manifestations. However, a heterozygous status may also contribute to carcinogenesis. +/-Men1 mutant mice develop prostate cancer due to drastically reduced expression of menin in the carcinogenic tissue. Despite this, only some tumor tissues demonstrate LOH.11 Furthermore, a study using DNA sequencing of formalin-fixed human prostate cancer biopsies revealed a missense mutation of Men1.12Finally, neuroendocrine tumors from MEN1 patients express genes, such as FGF9, which can stimulate the growth of pre-existing prostate cancer.13 Similar observations have been reported in women with breast cancer, although the role of menin in these cases seems more complicated. In sporadic breast cancer, Men1 mRNA is overexpressed and probably stimulates tumor proliferation by regulation of estrogen receptor alpha activity. Interestingly, a Men1 somatic mutation is rarely detected in these tumors. In contrast, females with MEN1 syndrome, who have reduced Men1 mRNA expression, demonstrate an unexpectedly increased risk of invasive breast cancer. This can be partially explained by the higher frequency of LOH in these cases compared to sporadic breast cancer. However, just one third of these tumors demonstrate LOH, although in most of them the nuclear localisation of menin is severely reduced.4,14,15 This indicates a more complex mechanistic pathway for menin, which has not been completely described as yet. To conclude, it seems that menin has a general role in carcinogenesis, including functions in non-endocrine organs that may previously have been underestimated.
A second important question is if the tumor manifestation or the course of the disease depends on the type of mutation. Previous studies have not shown a genotype-phenotype association.16 However, novel studies dispute this point. Firstly, the type of Men1 mutation can lead either to a clinically overt MEN1 syndrome or to milder clinical conditions. For example, familial isolated hyperparathyroidism (FIHP) is characterised by the development of PHPT alone and is associated with specific Men1 mutations, often missense (in approximately 40% of the cases) and only occasionally nonsense or frameshift (30% of the cases) mutations. Conversely, in MEN1 syndrome with a classical phenotype, a nonsense or frameshift mutation, which leads to a truncated and consequently inactivate protein, is present in 65% of cases.1 Although the reported mutation here is a missense one, the family meets the criteria for MEN1 syndrome according to the international guidelines.1 Subject 1 has developed two typical MEN1 manifestations (PHPT, pituitary adenoma), while subjects 2 and 3 (first degree relatives of subject 1 have developed PHPT. Finally, subject 4 carries the same mutation and was diagnosed with PHPT at the first screening for MEN1 at the age of 21, relatively early for FIHP. As subject 1 was the first to be diagnosed with MEN1, the late diagnosis of PHPT at the age of 72 in this subject can be explained by the lack of early screenings. The type of mutation can often affect the course of the disease. For example, patients with Men1-mutations resulting in loss of interaction with the checkpoint kinase 1-interacting domain have a higher risk of malignant pancreatic neuroendocrine neoplasia with aggressive course of disease and higher rates of disease-related death.17 Moreover, patients with mutations affecting the JunD-interacting domain have an increased risk of death secondary to a typical MEN1 tumor, demanding a more aggressive therapeutic approach.18 The novel Men1 mutation reported here is located in codon 38 of exon 2. Exon 1 is not translated and most of the mutations in exon 2 are frameshift or nonsense mutations leading to truncated menin. However, the current mutation leads to a change in one amino acid (from serine to cysteine). The first 40 amino-acids at the N-terminal end of menin are responsible for interactions with the transcription factor JunD. This novel mutation may therefore affect the function of the Menin-JunD complex. Interestingly, JunD has been linked to progression of prostate cancer due to overproduction of reactive oxygen species in cancer cells.19 Taking into consideration the family history and possible associations between MEN1 and prostate carcinogenesis, we recommended urological evaluation and annual biochemical screening (PSA) to patient 2.
Regarding the other two atypical tumor entities (renal cell carcinoma and papillary thyroid cancer), there are few relevant reports in other MEN1-families.20 In one case, a genetic analysis of papillary cancer tissue did not show LOH in a MEN1-patient, suggesting that the papillary cancer was not etiologically associated with the Men1 mutation.20 In our case, there was also no LOH observed in the tissue sample from subject 1. Since there are no in vitro, animal or human studies associating germline mutations of Men1 with the development of papillary thyroid cancer, we consider this manifestation in subject 1 a random observation. As patient 2 had already undergone a total thyroidectomy because of the cold thyroid nodule, further controls with ultrasonography or biomarkers were not necessary. Finally, the renal cell carcinoma detected in patient 1 is a rarity in MEN1 families. Since both patients receive, additionally to the biochemical screening, annual abdominal MRIs due to the increased risk of gastrointestinal tumors, we have not recommended an additional diagnostic work-up. It was judged that only minor diagnostic adaptations were necessary.
CONCLUSION
We describe a new germline mutation of Men1 in a german family, which is associated with an atypical phenotype. Since the role of menin as a tumor regulator in many organs remains to be established, it is difficult to distinguish between random observations and etiological relations in MEN1 families with atypical tumors. For this reason, situational adaptations of the recommended diagnostic procedure and follow-up should be considered. Future studies should focus on the potential effect of Men1 mutations on the development and progression of common but in MEN1 patients rarely described non-endocrine tumors.
DISCLOSURE STATEMENT
The authors have nothing to disclose.
CONCENT
Written informed consent for the genetic testing and the publication was obtained from all subjects.
REFERENCES
1. Thakker RV, Newey PJ, Walls GV, et al, 2012 Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 97: 2990-3011.
2. Koch CA, Pacak K, Chrousos GP, 2002 The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J Clin Endocrinol Metab 87: 5367-5384.
3. Cavalli T, Giudici F, Nesi G, et al, 2014 Sarcomatoid carcinoma of the kidney in a MEN1 patient: case report and genetic profile. Endocr J 61: 781-787.
4. Dreijerink KM, Goudet P, Burgess JR, Valk GD, 2014 Breast-cancer predisposition in multiple endocrine neoplasia type 1. N Engl J Med 371: 583-584.
5. Gao SB, Feng ZJ, Xu B, et al, 2011 Menin represses malignant phenotypes of melanoma through regulating multiple pathways. J Cell Mol Med 15: 2353-2363.
6. Gao SB, Xu B, Ding LH, et al, 2014 The functional and mechanistic relatedness of EZH2 and menin in hepatocellular carcinoma. J Hepatol 61: 832-839.
7. Harding B, Lemos MC, Reed AA, et al, 2009 Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia. Endocr Relat Cancer 16: 1313-1327.
8. Ren F, Xu HW, Hu Y, et al, 2012 Expression and subcellular localization of menin in human cancer cells. Exp Ther Med 3: 1087-1091.
9. Yang YQ, Qi J, Xu JQ, Hao P, 2014 MicroRNA-142-3p, a novel target of tumor suppressor menin, inhibits osteosarcoma cell proliferation by down-regulation of FASN. Tumour Biol 35: 10287-10293.
10. Fang M, Xia F, Mahalingam M, Virbasius CM, Wajapeyee N, Green MR, 2013 MEN1 is a melanoma tumor suppressor that preserves genomic integrity by stimulating transcription of genes that promote homologous recombination-directed DNA repair. Mol Cell Biol 33: 2635-2647.
11. Seigne C, Fontaniere S, Carreira C, et al, 2010 Characterisation of prostate cancer lesions in heterozygous Men1 mutant mice. BMC Cancer 10: 395.
12. Manson-Bahr D, Ball R, Gundem G, et al, 2015 Mutation detection in formalin-fixed prostate cancer biopsies taken at the time of diagnosis using next-generation DNA sequencing. J Clin Pathol 68: 212-217.
13. Dilley WG, Kalyanaraman S, Verma S, Cobb JP, Laramie JM, Lairmore TC, 2005 Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol Cancer 4: 9.
14. Brennan P, 2015 Breast cancer risk in MEN1 - a cancer genetics perspective. Clin Endocrinol (Oxf) 82: 327-329.
15. Dreijerink KM, Valk GD, 2015 Reply to: Breast cancer risk in MEN1 - a cancer genetics perspective. Clin Endocrinol (Oxf) 83: 141.
16. Lemos MC, Thakker RV, 2008 Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29: 22-32.
17. Bartsch DK, Slater EP, Albers M, et al, 2014 Higher risk of aggressive pancreatic neuroendocrine tumors in MEN1 patients with MEN1 mutations affecting the CHES1 interacting MENIN domain. J Clin Endocrinol Metab 99: E2387-291.
18. Thevenon J, Bourredjem A, Faivre L, et al, 2013 Higher risk of death among MEN1 patients with mutations in the JunD interacting domain: a Groupe d’etude des Tumeurs Endocrines (GTE) cohort study. Hum Mol Genet 22: 1940-1948.
19. Mehraein-Ghomi F, Kegel SJ, Church DR, et al, 2014 Targeting androgen receptor and JunD interaction for prevention of prostate cancer progression. Prostate 74: 792-803.
20. Desai D, McPherson LA, Higgins JP, Weigel RJ, 2001 Genetic analysis of a papillary thyroid carcinoma in a patient with MEN1. Ann Surg Oncol 8: 342-346.
Address for correspondence:
Dr. Nikolaos Perakakis, MD, Division of Endocrinology and Diabetology, Department of Internal Medicine II, University Hospital
of Freiburg, 55 Hugstetter Str., 79106 Freiburg, Germany; Tel.: +4976127034200, Fax: +4976127034130,
e-mail: nikolaos.perakakis@uniklinik-freiburg.de
Received: 28-05-2015, Accepted: 09-09-2015