1Medical Genetics Research Center, Department of Medical Genetics, School of Medicine, Mashhad University of Medical Sciences, 2Division of Human Genetics, Immunology Research Center, Avicena Research Institute, Mashhad University of Medical Sciences, 3Endocrine Research Center, Imam Reza/Ghaem Hospital, School of Medicine, Mashhad University of Medical Sciences; Mashhad, Iran
PURPOSE: Multiple Endocrine
Neoplasia type 2A (MEN2A) is a complex autosomal dominant inherited syndrome
characterized by medullary thyroid carcinoma (MTC), pheochromocytoma and
primary parathyroid hyperplasia. In patients with only one or two clinical
features, identification of a germ line RET (REarranged in Transfection)
mutation is required to make the diagnosis and initiate genetic counseling.
METHODS: We analyzed blood DNA from three Iranian families with three
generations of MEN2A including 20 affected individuals with MTC and four with
pheochromocytoma. RET hotspots were amplified in probands and sequenced
for mutation detection.
RESULT: The causative mutation in all families was
found to be the Cys634Tyr missense substitution. The presence of a functional
SNP resulting in Gly691Ser was also detected in exon 11 of 15 affected cases.
Four patients showed both of these RET variations.
CONCLUSION: Our study
shows that the Cys634Tyr missense substitution and the Gly691Ser polymorphism
are recurrent in Iranian patients, since our families are unrelated. All
asymptomatic carriers of the Cys634Tyr high-risk activating mutation were
referred for prophylactic thyroidectomy.
Cys634Tyr mutation, Gly691Ser polymorphism, Multiple Endocrine Neoplasia type 2A, RET proto-oncogene
INTRODUCTION
The human RET proto-oncogene maps to 10q11.2 and encodes a tyrosine kinase transmembrane receptor.1 The RET protein has three domains including an N-terminal extracellular domain that is a ligand for the GDNF neurotropic molecule, a hydrophobic transmembrane domain, and an intercellular tyrosine kinase domain.2 The binding of the GDNF family ligand to RET triggers homo-dimerization of RET and a transformational change in the RET intra-cytoplasmic domain.3 Germline mutations of the RET proto-oncogene cause the dominant inheritance of multiple endocrine neoplasia type 2A (MEN2A), MEN2B, and familial medullary thyroid carcinoma syndromes. Classical MEN2A is the most common MEN2A variant and in 95% of patients RET germline mutations occur in codons 609, 611, 618, or 620 of exon 10, or in codon 634 of exon 11. Virtually all patients with classical MEN2A present with medullary thyroid carcinoma (MTC) while fewer develop pheochromocytoma (PHEO) and primary parathyroid hyperplasia, the frequency of each depending on the specific RET mutation.4 According to the categorization of the American Thyroid Association, the ATA-HST “highest risk” includes patients with MEN2B and the RET codon M918T mutation, the ATA-H “high” category includes patients with RET codon C634 mutations, and the ATA-MOD “moderate risk” category includes patients with RET codon mutations other than M918T and C634.5
Among all thyroid tumors, MTC has a frequency of 5-10% and only 25% of these cases are categorized as familial type. Since thyroid tumors represent only 1% of all human cancer types, the incidence of MEN2 syndromes is estimated to be very low with approximately 500 to 1.000 MEN2 families existing worldwide.6 Owing to the rarity of the MEN2 syndromes, the prevalence of different RET mutations in distinct geographic areas is not well defined.7-9 As a rule, the clinical presentation of MEN2 syndromes will be more variable when the transforming activity of RET mutation is low.2,10,11 Genetic analysis of the RET proto-oncogene can be performed in those families affected by MEN2A and MEN2B, in cases of sporadic MTC or HSCR, and allows exact molecular diagnosis of the disease.12
To the best of our knowledge, this study is the first report of genetic screening of families of the Eastern region of Iran for identification of MEN2A families. We checked all mutation hotspots of the RET proto-oncogene in exons 11, 10, 13, and 8 by direct sequencing to produce reliable results.
METHODS
Family Selection and Ethics Statement
Three unrelated families who were affected by MEN2A (all from the Khorasan province, Iran) were included in this study after biochemical assays and clinical diagnosis by two endocrine specialists from the Endocrinology Research Center of Mashhad University of Medical Sciences (MUMS). The inclusion criteria for MEN2A phenotype was in line with the guidelines of the American Thyroid Association guidelines on MTC management.13 All the patients signed an informed consent to genetic analysis approved by the MUMS ethics committee according to the Declaration of Helsinki (1964). In cases of non-availability of patients, the required information was requested from at least two adult family members and hospital records were checked when available. Parents were asked to sign consents for children under the age of 15 years. Clinical features were evaluated over an average of 5 years of follow-up.
Amplification of RET hotspots
Genomic DNA was prepared from peripheral blood leukocytes by standard procedures. DNA samples were amplified from RET exons 8, 10, 11, and 13 by polymerase chain reaction (PCR) using a thermal cycler and specific primers. Nucleotide sequences of the primers were as follows: exon 8, 5′ TTGGGCACTAGCTGGACG 3′ and 5′ ACCTTCCCAAGTCCAGAGT 3′; exon 10, 5′ AGGCTGAGTGGGCTACGTCTG 3′ and 5′ GTTGAGACCTCTGTGGGGCT 3′; exon 11, 5′ ATGAGGCAGAGCATACGCAGCC 3′ and 5′ CTTGAAGGCATCCACGGAGACC 3′; exon 13, 5′ AACTTGGGCAAGGCGATGCA 3′ and 5′AGAACAGGGCTGTATGGAGC 3′.14 The PCR for the sequencing was performed in a volume of 50 µl containing 0.5 µM of each oligonucleotide primer, 50 ng of DNA, 1×PCR buffer, 250 µM dNTP, and 2.5 U of Taq polymerase using an automated thermal cycler (Techne, Flexigene, UK). The PCR was started with 5 minutes of pre-denaturation at 95°C, followed by 35 cycles of 40 seconds at 95°C, 30 seconds at 62°C, 55°C, 56°C, and 64°C for exons 8, 10, 11, and 13, respectively, then 40 seconds at 72°C; lastly, the procedure was completed with 10 minutes at 72°C for the final extension. Following PCR, the amplicon sizes were analyzed in 2% agarose gel and the products were visualized by green viewer staining. The PCR products were subjected to direct cycle sequencing and restriction enzyme analysis. The sequencing results were aligned to the RET reference sequence with Sequencher version 5.1 software.
Restriction fragment length polymorphism
The RFLP analyses of the RET cys634Tyr mutation was performed using RsaI restriction enzymes (Jena Bioscience, Germany) for genotyping of family members. Briefly, the restriction digestion was carried out at 37°C for 2 hours. Each reaction mixture contained 8 µl of the 333 base pair PCR product, 0.1 µl of RsaI (1 U), 2 µl of enzyme buffer, and 9.9 µl of distilled water to make a final volume of 20 µl for mutation identification. The digestion products were analyzed in 3% agarose gel stained with green viewer for RET gene fragments, respectively.
RESULTS
Among 20 patients belonging to three families with three generations of MEN2A, four (20 %) presented with PHEO. There were 11 females and 9 males with a mean age of 31 years. The diagnosis of PHEO was made after medullary thyroid carcinoma (n=3, 75%) and before MTC (n=1, 25 %). The family pedigrees are shown in Figures 1, 2, and 3.
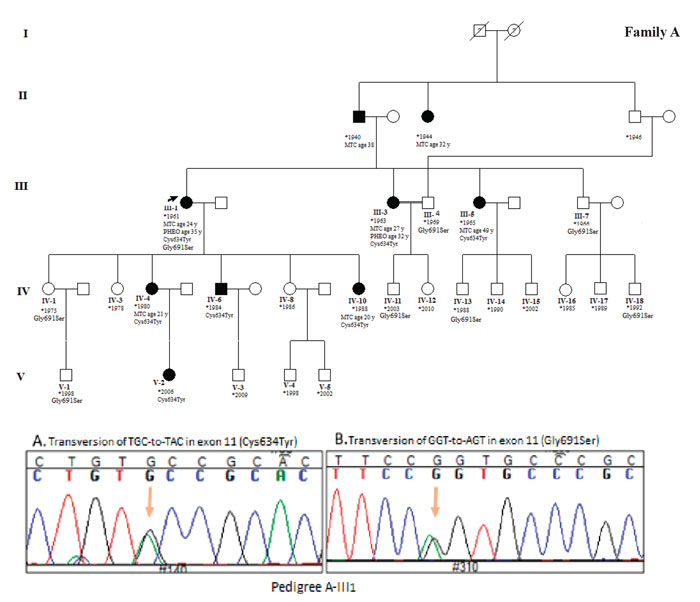
Figure 1. Pedigree of family A. Proband is shown by left arrow. All members with Roman numerals are genetically analyzed. Cases IV6 and V2 were recommended for prophylactic thyroidectomy. Sequencing results illustrated Cys634Tyr mutation and Gly691Ser polymorphism in exon 11 of proband.
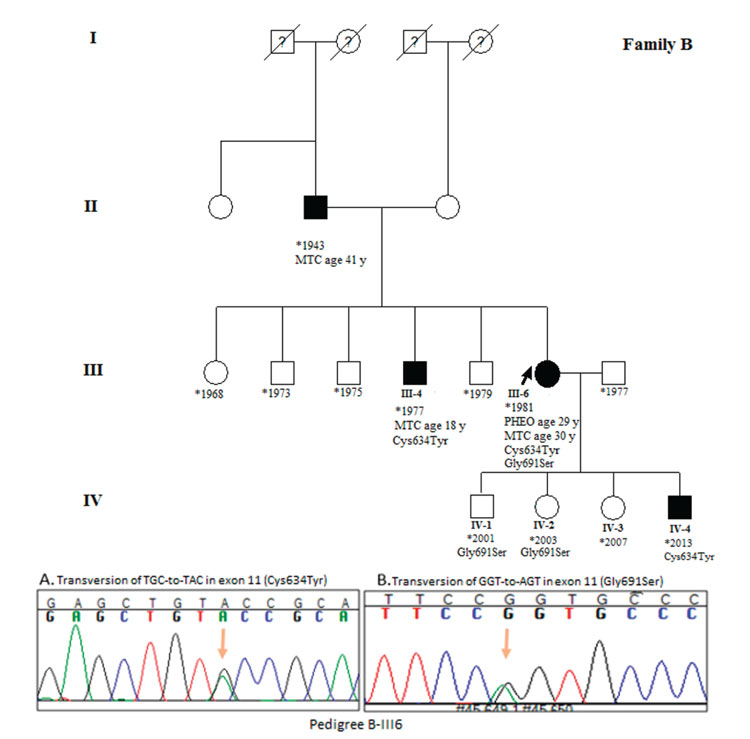
Figure 2. Pedigree of family A. Proband is shown by left arrow. All members with Roman numerals are genetically analyzed. Case IV4 was recommended for prophylactic thyroidectomy. Sequencing results illustrated Cys634Tyr mutation and Gly691Ser polymorphism in exon 11 of proband.
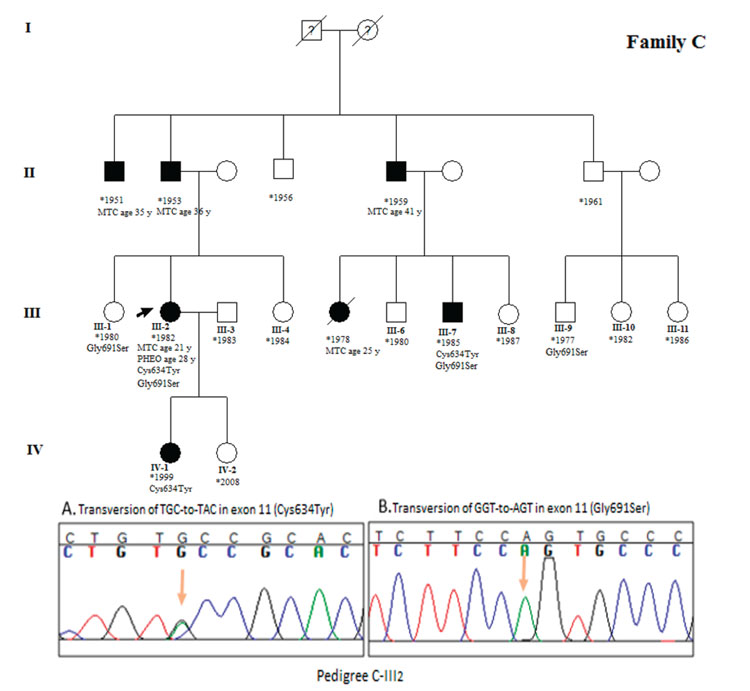
Figure 3. Pedigree of family C. Proband is shown by left arrow. All members with Roman numerals are genetically analyzed. Cases III7 and IV4 were recommended for prophylactic thyroidectomy. Sequencing results illustrated Cys634Tyr mutation and Gly691Ser polymorphism in exon 11 of proband.
Blood samples of 42 family members were collected and DNA extracted for genetic analysis. RET hotspots including exon 8, 10, 11, and 13 were amplified in 3 index cases (A-III1, B-III6 and C-III2) and products were analyzed in 2% agarose gel stained with green viewer. DNA sequence analysis for 4 exons of the RET gene revealed a transversion of TGC-to-TAC at codon 634 of exon 11 resulting in cysteine to tyrosine amino acid change. Additionally, we detected a Gly691Ser polymorphism in exon 11 of all the probands.
We then studied exon 11 in the family members using both RFLP and direct sequencing. RFLP was designed for Cys634Tyr mutation detection using RsaI endonuclease. RsaI cut the wild sequence to 308 and 25 base pairs where it cut the mutated sequence into two locations and made three bands of 230, 77, and 25 base pairs. Results from 3% agarose gel are shown in Figure 4.
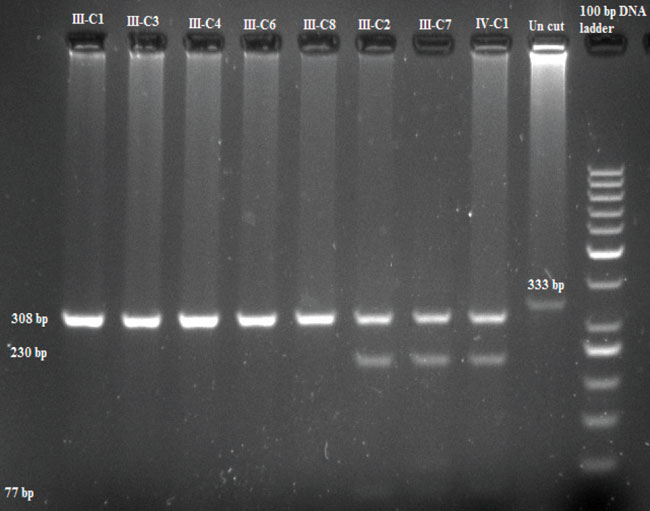
Figure 4. RFLP results of family C using RsaI endonuclease.
The sequencing of exon 11 in the probands and the family members who showed a mutation confirmed the result obtained from the RFLP test. According to the sequencing result, in family A, cases A-IV6 and A-V2 were asymptotic Cys634Tyr mutation carriers who were unaware of their condition. Genetic counseling was given to these cases. Case IV-A6 showed only a palpable thyroid. We found Gly691Ser polymorphisms in cases A-III1, A-III7, A-III9, A-IV1, A-IV11, A-IV13, A-IV18, and A-V1. Mild goiter and hypertension is clinical features of cases A-IV1, A-IV11, and A-IV18, while cases A-IV13 and A-V1 showed only a palpable thyroid. The proband of family A is a 53 year-old woman who has both a Cys634Tyr mutation and the Gly691Ser polymorphism in exon 11. She presented MTC at the age of 24 and PHEO at the age of 35. In family B, case number B-IV4 is a 6 month-old asymptomatic son carrying the Cys634Tyr mutation. The other two children of the B-III6 index case including B-IV1 and B-IV2 are carriers of the Gly691Ser polymorphism. The proband is a 33 year-old woman who presented with PHEO one year before MTC at the age of 29. In family C, three carriers of the Cys634Tyr mutation including two asymptomatic cases (C-III7 and C-IV1) were found. Case number C-III7 exhibited hypertension and mild goiter. The Gly691Ser polymorphism was detected in cases C-III1, C-III2, C-III7, and C-III9. Both C-III2 and C-III7 cases showed co-presentation of Cys634Tyr and Gly691Ser substitutions. One known silent mutation (rs1800861) was also detected in exon 13 of the members of families A and C.
Among a total of 42 genetically analyzed cases, 13 showed the Cys634Tyr high-risk activating mutation, 15 showed the Gly691Ser polymorphism, and four cases showed both of them. Five asymptomatic Cys634Tyr mutation carriers including A-IV6, A-V2, B-IV4, C-III7, and C-IV1 were referred for prophylactic thyroidectomy. All the sequencing results in the three index cases and five mutation carriers were repeated by reverse primer reading.
DISCUSSION
Mutations in the RET proto-oncogene have been implicated in the malignant transformation of parafollicular cells (C cells) of the thyroid that originate from the neural crest during embryogenesis.15 Germline mutations in RET cause Hirschsprung disease, which is a congenital defect of the enteric nervous system in the hindgut, and MEN2. Mutations converting the RET proto-oncogene into a dominant transforming gene is responsible for tumor components of MEN2A.16 Allelic imbalance through a tandem duplication and the resulting amplification of mutated RET has been proposed as a possible mechanism of tumor initiation in some patients with MEN2A-related MTC and pheochromocytoma.17-19
In most cases of MEN2A, thyroid carcinoma is the first clinical feature, which makes it difficult to diagnose between familial MTC cases and MEN2A without genetic testing. An Iranian MEN2A family first studied by Dr. Moosavi et al in 1992 displayed only MTC in 15 affected family members for many years.20
The most prevalent germline mutation encountered in patients with MEN2A is within codon 634 in the Cysteine-rich domain. Cases with the Cys634Arg mutation show more frequent and early metastases when compared to cases with Cys634Tyr.21-23 Mutations in codon 634 have a higher potency of cell neoplastic transformation than those in codons 609 and 611. This finding may be due to modulations in expression of mature RET-encoded protein receptors in the cell membrane.24,25 Thus, most patients with MEN2 who harbor RET 634 mutations will have adrenal and parathyroid tumors in addition to thyroid tumors, whereas patients with 609 or 611 mutations will present only with thyroid cancer.26 Codon-specific RET mutations may also have roles in tissue-specific sensitivity. Therefore, there is a high sensitivity in thyroid tissue, intermediate in the adrenals, and low in parathyroid glands.27 RET polymorphisms and haplotypes are believed to be genetic modifiers and might be associated with an increased relative risk for the development of disorders. The role of the RET variant allele G691S in MTC has been controversial. The G691S missense polymorphism might alter the function of the protein through creation of a new phosphorylation site affecting downstream signaling or changing the secondary structure of RET. It may be possible that the G691S variant has a role in disruption of topological chromatin domains if it presents a gene-enhancer activity.28 A recent meta-analysis concluded that the G691S increases the risk of several cancer types, including MTC, via a recessive mechanism of action.29 Some evidence shows that the RET variant G691S is a disease modifier in sporadic MTC.30
It has been suggested that the age of onset of MEN2A can be modified by RET G691S and S904S polymorphisms.31 Results from a study of Borrello M. (2011) demonstrated that, although RET-G691S is not oncogenic, it enhances the transforming activity of the RET-K666E mutant, hence suggesting a modifier role for this functional polymorphism.32 Because the transition from C-cell hyperplasia to node-negative and ultimately node-positive MTC takes time, the aforementioned histopathological phases are separated by time intervals.10,11 The time lag between malignant transformation and tumor cell spread represents a “window of opportunity” for surgical intervention before the tumor extends beyond the confines of the thyroid gland rendering it harder to cure. For carriers of RET mutations in codon 634, this time interval has been estimated to be around 6.6 years based on the mean age difference between patients with node-negative (10.2 years) and patients with node-positive thyroid cancer (17.1 years).10 Effective clinical interventions after appropriate genetic counseling are available for prevention through prophylactic thyroidectomy, and, if needed, adrenalectomy and parathyroidectomy.33 In conclusion, our study showed the presence of the Cys634Tyr mutation and Gly691Ser polymorphism in three Iranian families. Five asymptotic cases with the Cys634Tyr were unaware of their condition and were referred for prophylactic thyroidectomy.
ACKNOWLEDGEMENTS
This research was funded by Mashhad University of Medical Sciences. We are grateful for the generous assistance of Farzaneh Mohebi, nursing head of Imam Reza Hospital. We also thank all patients and family members for their patience and confidence.
CONFLICT OF INTEREST
We declare that we have no conflict of interest.
ETHICAL APPROVAL
All procedures performed in this study were in accordance with the ethical standards of the ethical committee of Mashhad University of Medical Sciences and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
REFERENCES
1. Takahashi M, Buma Y, Iwamoto T, Inaguma Y, Ikeda H, Hiai H, 1988 Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene 3: 571-578.
2. Machens A, Lorenz K, Dralle H, 2009 Constitutive RET tyrosine kinase activation in hereditary medullary thyroid cancer: clinical opportunities. J Ιntern Med 266: 114-125.
3. Airaksinen MS, Saarma M, 2002 The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: 383-394.
4. Imai T, Uchino S, Okamoto T, et al, 2013 High penetrance of pheochromocytoma in multiple endocrine neoplasia 2 caused by germ line RET codon 634 mutation in Japanese patients. Eur J Endocrinol 168: 683-687.
5. Wells SA Jr, Asa SL, Dralle H, 2015 Revised american thyroid association guidelines for the management of medullary thyroid carcinoma .Thyroid 25: 567-610.
6. Keiser HR, Beaven MA, Doppman J, Wells S Jr., Buja LM, 1973 Sipple’s syndrome: medullary thyroid carcinoma, pheochromocytoma, and parathyroid disease. Studies in a large family. NIH conference. Ann Intern Med 78: 561-579.
7. Pinna G, Orgiana G, Riola A, et al, 2007 RET proto-oncogene in Sardinia: V804M is the most frequent mutation and may be associated with FMTC/MEN-2A phenotype. Thyroid 17: 101-104.
8. Berndt I, Reuter M, Saller B, et al, 1998 A new hot spot for mutations in the ret protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 83: 770-774.
9. Machens A, Dralle H, 2008 Familial prevalence and age of RET germline mutations: implications for screening. Clinical endocrinol 69: 81-87.
10. Machens A, Niccoli-Sire P, Hoegel J, et al, 2003 Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 349: 1517-1525.
11. Machens A, Lorenz K, Dralle H, 2009 Individualization of lymph node dissection in RET (rearranged during transfection) carriers at risk for medullary thyroid cancer: value of pretherapeutic calcitonin levels. Ann Surg 250: 305-310.
12. Martucciello G, Lerone M, Bricco L, et al, 2012 Multiple endocrine neoplasias type 2B and RET proto-oncogene. Ital J Pediatr 38: 9.
13. American Thyroid Association Guidelines Task F, Kloos RT, Eng C, Evans DB, et al, 2009 Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19: 565-612.
14. Zhou Y, Zhao Y, Cui B, et al, 2007 RET proto-oncogene mutations are restricted to codons 634 and 918 in mainland Chinese families with MEN2A and MEN2B. Clin Endocrinol(Oxf) 67: 570-576.
15. Manie S, Santoro M, Fusco A, Billaud M, 2001 The RET receptor: function in development and dysfunction in congenital malformation. Trends Genet 17: 580-589.
16. Landsvater RM, de Wit MJ, Zewald RA, et al, 1996 Somatic mutations of the RET proto-oncogene are not required for tumor development in multiple endocrine neoplasia type 2 (MEN 2) gene carriers. Cancer Res 56: 4853-4855.
17. Huang SC, Koch CA, Vortmeyer AO, et al, 2000 Duplication of the mutant RET allele in trisomy 10 or loss of the wild-type allele in multiple endocrine neoplasia type 2-associated pheochromocytomas. Cancer Res 60: 6223-6226.
18. Koch CA, Huang SC, Moley JF, et al, 2001 Allelic imbalance of the mutant and wild-type RET allele in MEN 2A-associated medullary thyroid carcinoma. Oncogene 20: 7809-7811.
19. Huang SC, Torres-Cruz J, Pack SD, et al, 2003 Amplification and overexpression of mutant RET in multiple endocrine neoplasia type 2-associated medullary thyroid carcinoma. J Clin Endocrinol Metab 88: 459-463.
20. Mosavi Z, Abdinejad A 1992 Familial Medullary Thyroid Carcinoma. Report of 15 cases in a family members. The Second International Congress on Endocrine Disorders; Tehran-Iran.
21. Brandi ML, Gagel RF, Angeli A, et al, 2001 Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86: 5658-5671.
22. Hoff AO, Cote GJ, Gagel RF, 2000 Multiple endocrine neoplasias. Annu Rev Physiol 62: 377-411.
23. Niccoli-Sire P, Murat A, Baudin E, et al, 1999 Early or prophylactic thyroidectomy in MEN 2/FMTC gene carriers: results in 71 thyroidectomized patients. The French Calcitonin Tumours Study Group (GETC). Eur J Endocrinolo 141: 468-474.
24. Eng C, Smith DP, Mulligan LM, et al, 1994 Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet 3: 237-241.
25. Punales MK, Graf H, Gross JL, Maia AL, 2003 RET codon 634 mutations in multiple endocrine neoplasia type 2: variable clinical features and clinical outcome. J Clin Endocrinol Metab 88: 2644-2649.
26. Toledo SP, dos Santos MA, Toledo Rde A, Lourenco DM Jr, 2006 Impact of RET proto-oncogene analysis on the clinical management of multiple endocrine neoplasia type 2. Clinics (Sao paulo) 61: 59-70.
27. Marx SJ, 2005 Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat Rev Cancer 5: 367-375.
28. Lupianez DG, Kraft K, Heinrich V, et al, 2015 Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161: 1012-1025.
29. Lantieri F, Caroli F, Ceccherini I, Griseri P, 2013 The involvement of the RET variant G691S in medullary thyroid carcinoma enlightened by a meta-analysis study. Int J Cancer 132: 2808-2819.
30. Machens A, Frank-Raue K, Lorenz K, Rondot S, Raue F, Dralle H, 2012 Clinical relevance of RET variants G691S, L769L, S836S and S904S to sporadic medullary thyroid cancer. Clin Endocrinol 76: 691-697.
31. Robledo M, Gil L, Pollan M, et al, 2003 Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res 63: 1814-1817.
32. Borrello MG, Aiello A, Peissel B, et al, 2011 Functional characterization of the MTC-associated germline RET-K666E mutation: evidence of oncogenic potential enhanced by the G691S polymorphism. Endocr Relat Cancer 18: 519-527.
33. Machens A, Lorenz K, Sekulla C, et al, 2013 Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol 168: 307-314.
Address for correspondence:
Zohreh Mosavi, M.D., Head, Endocrine Research Center, Imam Reza/Ghaem Hospital, School of Medicine, Mashhad University of Medical Sciences; Vakil-Abad Street, Postal code: 0888978421 Mashhad, Iran; Tel.: +98 5138012976, E-mail: MosaviZ@mums.ac.ir
Received:16-04-2015, Accepted:30-06-2015