1Paediatric Endocrinology Unit, Department of Health Sciences, 2Genetics and Molecular Medicine Unit; Anna Meyer Children’s University Hospital, Florence, Italy
OBJECTIVE: SOX3 is located
on the long arm of the X chromosome (Xq27.1) and both the under- and
over-expression of this gene have been reported in cases of hypopituitarism
with or without intellectual disabilities. Nevertheless, only a few cases have
as yet been extensively described.
DESIGN: A 3-year 11 month-old male was
brought in for growth failure (height -2.4 SDS). The patient was born at term
of a second uneventful pregnancy by caesarean section for podalic presentation:
the birth weight (0.1 SDS), length (0.4 SDS), and head circumference (-0.3 SDS)
were normal. Neurodevelopmental delays and ocular motor dyspraxia had been
noted since 6 months of age. The endocrinological evaluation showed a very low
IGF-I concentration (44 μg/L). The thyroid hormone level was normal and coeliac
disease markers were negative. Bone age was considerably delayed. Target height
was normal (0.5 SDS).
RESULTS: Growth hormone stimulation tests were compatible
with a classic GHD, while a brain MRI disclosed a pituitary hypoplasia with
ectopic neurohypophysis. rhGH treatment was then begun and the auxological
follow-up showed a good response. At the age of 9 yrs, the height was 0.3 SDS,
the weight was 0.1 SDS, and the pubertal evaluation was PH1 AH1 T2 ml
bilaterally. Due to the presence of neuromotor delays and MRI abnormalities, a
genetic evaluation was conducted and an array-CGH of the patient’s DNA
discovered an Xq26.3-27.3 duplication comprising the SOX3 gene.
CONCLUSIONS: SOX3 involvement should be considered in a male with short
stature due to GH deficiency associated with intellectual disability.
Dyspraxia, Growth Hormone, Growth Hormone Deficiency, Intellectual Disability Short stature, SOX3
INTRODUCTION
Pituitary gland development and function depend on the sequential temporal and spatial expression of multiple transcription factor genes, such as POU1F1 (POU class 1 homeobox 1; OMIM 173110), PROP1 (prophet of PIT1; OMIM 601538), HESX1 (homeobox gene expressed in ES cells; OMIM 601802), LHX3 (LIM homeobox gene 3; OMIM 600577), LHX4 (LIM homeobox gene 4; OMIM 602146), SOX3 (SRY-related HMG-box gene 3; OMIM 313430), and OTX2 (orthodenticle homolog 2; OMIM 600037).1-3
Congenital hypopituitarism (CH), a defect that is characterised by a deficiency in one or more pituitary hormones and is not rare, may be caused by mutations in any of these genes.4
CH manifests either as an isolated hormone deficiency, the most common being isolated growth hormone deficiency (IGHD) or as multiple pituitary hormone involvement [combined pituitary hormone deficiencies (CPHD)].1-3
The CH clinical features may be detected in the neonatal period or present later in life.4,5 Moreover, hormonal deficits may be associated with extra-pituitary defects affecting organs that are embryologically correlated.4,5
SOX3 is a single exon gene located on the long arm of the X chromosome (Xq27.1). SOX3 is a member of the SOX (SRY-related high mobility group box) family of transcription factors that is expressed in neuroepithelial progenitor and stem cells beginning in the earliest stages of embryogenesis.6,7 Sox/SOX genes have been recognised as key players in the regulation of embryogenesis and nervous system development; they encode transcription factors that act as key regulators in different developmental processes, such as gastrulation, neural induction, specification, and the differentiation of many cell types.8,9 SOX3 has also been implicated in the aetiology of a septo-optic dysplasia variant.3
The dysfunction of the SOX3 protein disturbs cellular processes that are required for cognitive and pituitary development.10 In fact, in human males, both the under- and over-expression of this gene lead to CPHD or IGHD and infundibular hypoplasia, an ectopic/undescended posterior pituitary and abnormalities of the corpus callosum with or without intellectual disability (ID).10-12
However, micro-duplications of SOX3 have been identified in only a few patients with IGHD or CPHD,10,13-16 frequently accompanied by poor endocrinological13,15,16 or clinical10,13 data.
CASE REPORT
The propositus was the second child of young, healthy, non-consanguineous parents of Italian origin. The target height was normal [0.5 standard deviation score 9SDS)]. After a miscarriage, the couple had a son with normal growth and neuropsychological development (the pedigree is illustrated in Figure 1).
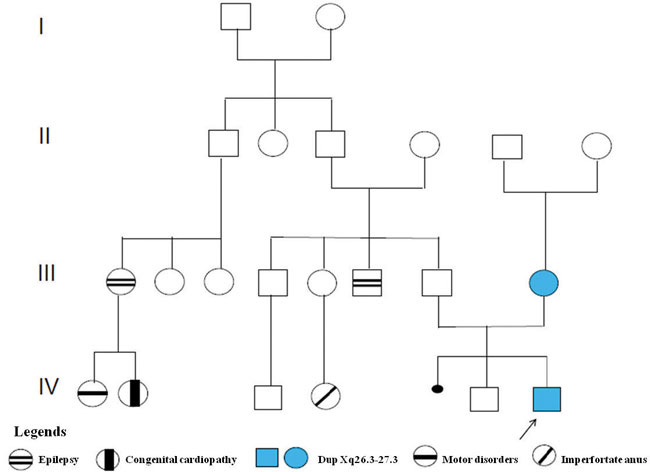
Figure 1. Pedigree of the family that was analysed in this study. Duplications were found in the proband and mother.
The patient was born by caesarean section for podalic presentation at term of the 3rd uneventful pregnancy. The birth weight was 3.030 kg (0.1 SDS), the length was 52 cm (0.4 SDS) and the head circumference was 34.8 cm (-0.3 SDS). The Apgar score was 9I-10V. Genital abnormalities were not observed, nor were hypoglycaemias.
During the first year of life, a mild developmental delay became evident: he sat at 8 months and walked independently at 22 months, while language started at 24 months. Intellectual disability was ascertained at the age of 2 yr and 6 mo: the developmental quotient (DQ) was 65. In the same period, an ophthalmologic examination was carried out: the fundus and visual acuity were normal, but a gaze-evoked horizontal nystagmus and ocular saccadic overshot were observed, leading to a suspicion of a diagnosis of ocular dyspraxia.
At 3 yr and 11 mo old, due to pronounced growth failure (height -2.4 SDS), an endocrinological evaluation of the child was conducted which revealed a very low IGF-I concentration (44 μg/L). An extensive endocrine work-up was performed: free-thyroxin [(FT4) 1.47 ng/dL, n.v. 0.86-2.12 ng/dL], thyroid-stimulating hormone [(TSH) 3.38 μUI/dL, n.v. 0.4-4.0 μUI/dL], cortisol (8.23 μg/dL, n.v. 5-25 μg/dL), adrenocorticotropic hormone [(ACTH) 50 ng/L, n.v. 09-52 ng/L), glucose (72 mg/dL, n.v. 55-110 mg/dL), and prolactin [(PRL) 86 mUI/ml] were in the normal range. The electrolyte, venous blood gas, haemoglobin, total protein, serum albumin, coagulation profile, calcium, phosphorous, vitamin D (25OHD), and parathyroid hormone (PTH) levels were also normal. The anti-tissue transglutaminase (tTG) was negative. Neuro-metabolic tests (plasma aminoacidogram, urine aminoacidogram, acylcarnitine profile analysis, and redox state) yielded normal results. The karyotype was 46,XY. A multiplex ligation-dependent probe amplification (MLPA) analysis and fragile X syndrome (FRAXA) testing also returned normal results.
Bone age was considerably delayed (2 yr and 1 mo at 3 yr and 11 mo of chronological age). A growth hormone stimulation test disclosed a classic GH deficiency (GH peak after clonidine 2.4 ng/mL; GH peak after arginine 2.1 ng/mL). An MRI revealed an anterior pituitary hypoplasia with ectopic neurohypophysis, corpus callosum hypogenesis, and incomplete myelination (Figure 2). The posterior fossa was significantly reduced.
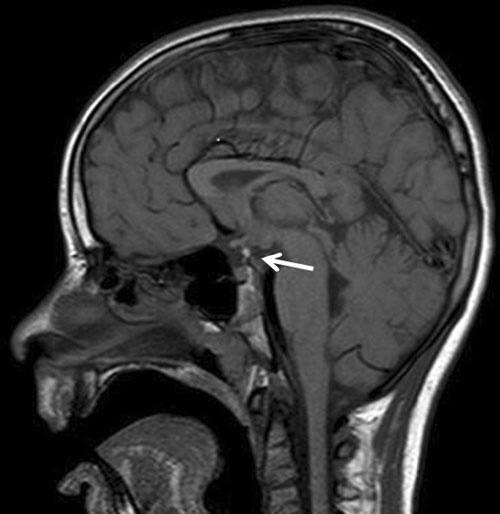
Figure 2. Sagittal MRI scan of the patient with SOX3 duplica- tion, showing pituitary hypoplasia, hypoplasia of the infundibu- lum, and an undescended/ectopic posterior pituitary. Note the hypogenesis of the corpus callosum.
Based on these findings, rhGH treatment was conducted (0.23 mg/kg per week subcutaneously). The auxological follow-up showed a strong positive response to the treatment with the standard deviation-growth velocity (SDS-GV) increasing remarkably during therapy (Figure 3).

Figure 3. Growth chart of the patient. The arrows indicate the age of onset for growth hormone treatment (black arrow).
At 9 yr and 9 mo, the height was 0.3 SDS, the weight was 0.1 SDS, and the pubertal evaluation was PH1 AH1 T2 ml bilaterally. Bone age remained considerably delayed (6 yr and 1 mo at 9 yr and 9 mo of chronological age). During follow-up, a periodic evaluation of the other adenohypophyseal hormones disclosed deficiency. At 6 yr and 8 mo of age, the FT4 was 1.56 ng/dL, the TSH was 3.01 μUI/dL, the cortisol was 11.45 μg/dL, the ACTH was 42 ng/L and the PRL was 69 mUI/ml.
Due to the presence of neuromotor delays and MRI abnormalities, a genetic evaluation was carried out. The patient’s DNA was analysed using array CGH (comparative genomic hybridisation). After obtaining informed consent, the genomic DNA was extracted from the leukocytes of the proband, i.e. both his parents and maternal grandparents according to standard procedures. Array CGH was performed using the Agilent 60k platform with a median resolution rate of nearly 100 kilobases (kb). Based on the physical mapping positions that were designated at the March 2006 assembly (NCBI36/hg18) of the UCSC Genome Browser, this analysis showed a duplication that involved the Xq26.3-27.3 region with an extension between 135,175,703 bp (first duplicated) and 142,971,531 bp (last duplicated) (Figure 4; Table 1): a 7.8 megabase (Mb) duplication was identified in Xq26.3 - 27.3 spanning more than 20 genes, among which the morbid genes were SOX3, FHL1 (four-and-a-half lim domains 1; OMIM 300163), CD40LG (CD40 antigen ligand; OMIM 300386), ARHGEF6 (rho guanine nucleotide exchange factor 6; OMIM 300267), ZIC3 (zinc finger protein of cerebellum 3; OMIM 300267), and F9 (coagulation factor IX; OMIM 300746). The same duplication was found in the DNA of the mother.
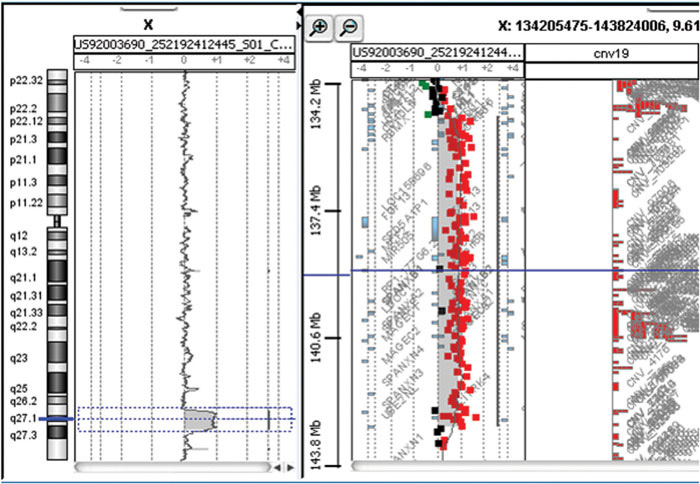
Figure 4. Array-CGH analysis showing a duplication that included Xq26.3-27.3 with the extension between 135,175,703 bp (first duplicated) and 142,971,531 bp (last duplicated).
DISCUSSION
We describe a new case of isolated GHD in a patient with the duplication Xq26.3-27.3 comprising the SOX3 gene. The growth failure was relatively severe, but a significant catch-up growth achieved after a long-term follow-up with rhGH treatment confirmed the diagnosis of GHD.
Additional pituitary deficiencies were not recorded, and an evaluation of a possible LH/FSH deficiency will be performed in the future.
GHD has been reported in most cases of SOX3 involvement (Tables 1 and 2).10,13,14,17-19 Recently, Takagi described a male patient with Kabuki syndrome due to a mutation in KMT2D (Lysine-Specific Methyltransferase 2D). KMT2D is involved in the majority of cases of Kabuki syndrome, a condition that is sometimes associated with GHD. As the patient also had CPHD, the authors analysed all of the coding exons and flanking introns of currently known genes responsible for CPHD by PCR-based sequencing, discovering a mutation in SOX3 consisting of a deletion in the polyalanine (PA) tracts of SOX3. This study provides additional evidence that SOX3 mutations must be looked for in hypopituitarism.
As documented by Woods et al, the over- and under-expression of SOX3 is associated with significant interfamilial phenotypic variability, which may be seen in many patients even with identically sized expansions.10 To the best of our knowledge, this case is the second described isolated case of GHD after the patient who was reported by Burkitt Wright.18 In fact, GHD is more frequently associated with TSH deficiencies, the exceptions being the cases published by Hol,14 Burkitt Wright,18 and ourselves; ACTH10,13,14,19 or gonadotropin10,13,17,18 deficiencies have been more rarely diagnosed, even though in many cases specific diagnostic tests were not carried out.16
Therefore, based on the evaluation of the various pituitary defects and molecular diagnoses (sequence variant and whole gene deletion or duplication), patients with duplication of SOX3 could present an IGHD without the involvement of additional adenohypophyseal hormones,10,14,15 more frequently with respect to patients with SOX3 sequence variants.10,17,19 Nevertheless, the cases that were reported by Hol et al14 and Woods et al10 with the duplication Xq26.3-27.3 also displayed in CPHD. Other subjects with SOX3 duplication that were described by Salomon et al20 have not been confirmed.21
Anterior pituitary hypoplasia, an absent stalk, and ectopic neurohypophysis are other useful findings that can support the diagnosis of CPHD or IGHD due to SOX3 sequence variants or whole gene deletions/duplications.22 However, some patients lack descriptions of their hypothalamic-pituitary anatomy,10,12-15 whereas others disclosed only partially the MRI characteristics that have been described as typical of SOX3 involvement. For example, in a case that was described by Woods et al,10 MRI abnormalities were absent, such as in the patient who was reported by Helle et al23 and one of the patients who was reported by Hol et al.14 Nevertheless, the case of Helle et al23 showed hyperphagia, most likely with a hypothalamic origin without other typical SOX3 involvement characteristics; this patient did not have hypopituitarism.
Available evidence demonstrates that either the over- or under-expression of SOX3 can result in the perturbation of pituitary and hypothalamic development.24 Altered SOX3 d osage may also be the causative mechanism for the X-linked hypopituitarism that is associated with infundibular hypoplasia, an ectopic/undescended posterior pituitary, and abnormalities of the corpus callosum (with or without ID).10,12,17,19 Therefore, the presence of IGHD or CPHD in males, in particular if presenting ID, may be a useful indicator of potential defects in SOX3.
ID is frequently reported in these patients. The degree of mental retardation and the characteristics vary among patients.12-15,23 Even though mental retardation is reported in the majority of patients with SOX3 duplications,13-15 this trait was not present in the cases that were documented by Woods et al10, which were characterised by a smaller duplication (685.6 kb in length). Although one of these patients exhibited hyperactivity, the absence of ID in some patients10,17,18 may result from different dosage effects.
Vertebrate embryonic stem cells express the Sox2 transcription factor, which, together with the closely related Sox1 and Sox3 proteins, forms the SoxB1 subgroup of the Sox protein family. First, Bergsland et al found that the genome-wide binding patterns of Sox2 and Sox3 in neural precursor cells (NPCs) overlap extensively, with 96% of the Sox2-bound sites also bound by Sox3.25 Therefore, a substantial number of the identified binding sites are part of brain-specific regulatory regions. Both high and low levels of Sox3 can deleteriously affect normal brain function and physiology. In fact, constitutively active Sox3 leads to increased apoptosis.26
In conclusion, IGHD and CPHD are frequently reported characteristics in patients with SOX3 involvement. The association with mental retardation is also typically present, more frequently in SOX3 duplication than in mutations. In the case of males with mental retardation and postnatal growth failure due to IGHD or associated with CHPD, the involvement of SOX3 may be considered.
REFERENCES
1. Cohen LE, Radovick S, 2002 Molecular basis of combined pituitary hormone deficiencies. Endocr Rev 23: 431-442.
2. Dateki S, Kosaka K, Hasegawa K, et al, 2010 Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J Clin Endocrinol Metab 95: 756-764.
3. Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT, 2009 Genetic regulation of pituitary gland development in human and mouse. Endocr Rev 30: 790-829.
4. Kelberman D, Dattani MT, 2007 Hypothalamic and pituitary development: novel insights into the aetiology. Eur J Endocrinol 157: Suppl 1: 3-14.
5. Dattani MT, 2005 Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clin Endocrinol (Oxf) 63: 121-130.
6. Collignon J, Sockanathan S, Hacker A, et al, 1996 A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development 122: 509-520.
7. Kamachi Y, Uchikawa M, Kondoh H, 2000 Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet 16: 182-187.
8. Pevny LH, Lovell-Badge R, 1997 Sox genes find their feet. Curr Opin Genet Dev 7: 338-344.
9. Lefebvre V, Dumitriu B, Penzo-Méndez A, Han Y, Pallavi B, 2007 Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int J Biochem Cell Biol 39: 2195-2214.
10. Woods KS, Cundall M, Turton J, et al, 2005 Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet 76: 833-849.
11. Solomon NM, Ross SA, Morgan T, et al, 2004 Array comparative genomic hybridisation analysis of boys with X linked hypopituitarism identifies a 3.9 Mb duplicated critical region at Xq27 containing SOX3. J Med Genet 41: 669-678.
12. Laumonnier F, Ronce N, Hamel BC, et al, 2002 Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet 71: 1450-1455.
13. Lagerström-Fermér M, Sundvall M, Johnsen E, et al, 1997 X-linked recessive panhypopituitarism associated with a regional duplication in Xq25-q26. Am J Hum Genet 60: 910-916.
14. Hol FA, Schepens MT, van Beersum SE, et al, 2000 Identification and characterization of an Xq26-q27 duplication in a family with spina bifida and panhypopituitarism suggests the involvement of two distinct genes. Genomics 69: 174-181.
15. Stankiewicz P, Thiele H, Schlicker M, et al, 2005 Duplication of Xq26.2-q27.1, including SOX3, in a mother and daughter with short stature and dyslalia. Am J Med Genet A 138: 11-17.
16. Moalem S, Babul-Hirji R, Stavropolous DJ, et al, 2012 XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet A 158A: 1759-1764.
17. Alatzoglou KS, Kelberman D, Dattani MT, 2009 The role of SOX proteins in normal pituitary development. J Endocrinol 200: 245-258.
18. Burkitt Wright EM, Perveen R, Clayton PE, et al, 2009 X-linked isolated growth hormone deficiency: expanding the phenotypic spectrum of SOX3 polyalanine tract expansions. Clin Dysmorphol 18: 218-221.
19. Takagi M, Ishii T, Torii C, Kosaki K, Hasegawa T, 2013 A novel mutation in SOX3 polyalanine tract: a case of kabuki syndrome with combined pituitary hormone deficiency harboring double mutations in MLL2 and SOX3. Pituitary Dec 18.
20. Solomon NM, Nouri S, Warne GL, Lagerström-Fermér M, Forrest SM, Thomas PQ, 2002 Increased gene dosage at Xq26-q27 is associated with X-linked hypopituitarism. Genomics 79: 553-559.
21. Solomon NM, Ross SA, Forrest SM, et al, 2007 Array comparative genomic hybridisation analysis of boys with X-linked hypopituitarism identifies a 3.9 Mb duplicated critical region at Xq27 containing SOX3. J Med Genet 44: e75.
22. Tillmann V, Tang VW, Price DA, Hughes DG, Wright NB, Clayton PE, 2000 Magnetic resonance imaging of the hypothalamic-pituitary axis in the diagnosis of growth hormone deficiency. J Pediatr Endocrinol Metab 13: 1577-1583.
23. Helle JR, Barøy T, Misceo D, Braaten Ø, Fannemel M, Frengen E, 2013 Hyperphagia, mild developmental delay but apparently no structural brain anomalies in a boy without SOX3 expression. Am J Med Genet A 161A: 1137-1142.
24. Hamel BC, Smits AP, Otten BJ, van den Helm B, Ropers HH, Mariman EC, 1996 Familial X-linked mental retardation and isolated growth hormone deficiency: clinical and molecular findings. Am J Med Genet 64: 35-41.
25. Bergsland M, Ramsköld D, Zaouter C, Klum S, Sandberg R, Muhr J, 2011 Sequentially acting Sox transcription factors in neural lineage development. Genes Dev 25: 2453-2464.
26. Archer TC, Jin J, Casey ES, 2011 Interaction of Sox1, Sox2, Sox3 and Oct4 during primary neurogenesis. Dev Biol 350: 429-440.
Address for correspondence:
Stefano Stagi, MD, Health Sciences Department, Anna Meyer Children’s University Hospital, Viale Pieraccini 24, 50139 Florence, Italy, Tel.: +39-055-5662622; Fax: +39-055-4221012; E-mail: stefano.stagi@yahoo.it
Received: 26-01-2014, Accepted: 16-06-2014