1Department of Endocrinology and Diabetes Center, General Hospital of Athens “G. Gennimatas”, Athens, Greece; 21st Department of Medicine, The University of Chicago, Medicine and Biological Sciences Division, Chicago, Illinois, USA; 3Department of Interventional Radiology, General Hospital of Athens “G. Gennimatas”, Athens, Greece
OBJECTIVE: Non-insulinoma pancreatogenous hypoglycemia syndrome (NIPHS) is one of the rare causes of endogenous hyperinsulinism. Its diagnosis is challenging and may require selective intraarterial calcium stimulation and concomitant hepatic vein sampling (SACVS). Impaired counterregulatory hormones’ production in response to hypoglycemia has been previously described in patients with diabetes, insulinoma and infancy hypoglycemia. We present a case of endogenous hyperinsulinism, secondary to NIPHS, with deficient cortisol response to hypoglycemia which resolved after diazoxide treatment.
DESIGN-RESULTS: A 43-year-old woman was admitted with recurrent episodes of registered fasting and postprandial hypoglycemia. Abdominal computed tomography, magnetic resonance imaging and endoscopic ultrasonography of the pancreas failed to reveal any lesion while SACVS sampling demonstrated a 5- to 8-fold increase in insulin levels in diverse parts of the pancreas. Counterregulatory hormones’ measurement revealed an attenuated cortisol response. Treatment with diazoxide resulted in disappearance of hypoglycemic episodes. Twelve months later, an insulin tolerance test was performed which revealed a normal cortisol response.
CONCLUSIONS: This report describes the first, to our knowledge, reported case in the literature of NIPHS with deficient cortisol response to hypoglycemia which resolved after diazoxide treatment. It is important for clinicians to include NIPHS in the differential diagnosis of hypoglycemia and identify possible impairment of counterregulatory hormones’ production.
Adrenaline, Cortisol, Counterregulatory hormones, Diazoxide, Endogenous hyperinsulinism GABA, Hypoglycemia, Hypoglycemia unawareness, Insulinoma, K-ATP channel, NIPHS, Non-pancreatogenous hypoglycemia syndrome
INTRODUCTION
Endogenous hyperinsulinism (EH) comprises a group of rare causes of hypoglycemia including insulinomas, non-insulinoma pancreatogenous hypoglycemia syndrome (NIPHS), autoimmune hypoglycemia and focal or diffuse islet cell hyperplasia in infants (congenital hyperinsulinism). Insulinomas are the most common cause of EH with an estimated incidence of 1 to 4 new cases per million patient-years.1,2 Rarely, they can be malignant or they can be encountered as part of MEN-1 syndrome or, even rarer, of neurofibromatosis type 1 and tuberous sclerosis.1,3-5 In 0.5-5% of patients, insulin excretion is derived from NIPHS.6,7
Patients with insulinomas are usually characterized by symptoms and signs related to fasting hypoglycemia and disproportionally high insulin levels in the presence of hypoglycemia. In the absence of typical registered hypoglycemic episodes, a prolonged supervised 72h fast is required. Frequently, insulinoma localization can be difficult and challenging due to their small size.8 Referral to specialized centers for endoscopic ultrasound and selective arterial calcium stimulation with hepatic vein sampling (SACVS) may be required. In most of the cases, surgical removal of the encapsulated tumor or part of the pancreas can be curative.
On the other hand, NIPHS is characterized by endogenous hyperinsulinemic hypoglycemia that is not caused by an insulinoma. The diagnosis should be suspected in patients with predominant postprandial hyperinsulinemic hypoglycemia, negative radiological localizing studies, and positive responses to SACVS in diverse parts of the pancreas. Τhe etiology of NIPHS remains unknown, while the histological exam shows beta-cell hypertrophy. The majority of cases are related to preceding bariatric surgery, suggesting a reactive process possibly due to glucagon-like peptide 1 (GLP-1)-induced islet cell proliferation and hyperplasia.9 Gene mutations encoding subunits (Kir6.2 and SUR1) of the pancreatic ATP-sensitive potassium (K-ATP) channel, which are encountered in cases of congenital hyperinsulinism, are lacking in NIPHS.9,10 Amelioration of symptoms occurs with partial or subtotal pancreatectomy, or with medical treatment with diazoxide or somatostatin analogues to block insulin release.
Regardless of the cause of hypoglycemia, counterregulatory hormones such as glucagon, adrenaline, noradrenaline, growth (GH), adrenocorticotropic hormone (ACTH) and cortisol are produced in increased amounts during a hypoglycemic episode in order to restore normal glucose levels. Normally, glucagon and epinephrine are secreted at the glycemic threshold level of approximately 3.8 mmol/L (68 mg/dL), GH secretion occurs at 3.7 mmol/L (66 mg/dL) and secretion of cortisol at 3.2 mmol/L (58 mg/dL).11 Recurrent and/or persistent episodes of hypoglycemia can lower the threshold of counterregulatory hormone production. Defects in counterregulatory hormone secretion can be present in patients with diabetes mellitus (DM), congenital hyperinsulinism and insulinoma who may present with attenuated adrenaline, and/or noradrenaline, GH and cortisol responses to hypoglycemia.12-16 Tumor enucleation or partial/subtotal pancreatectomy successfully relieves hypoglycemic symptoms and restores counterregulatory hormonal response in the majority of patients with insulinoma or NIPHS. However, restoration of defective hormonal response with pharmaceutical treatment in NIPHS patients has not been previously described.
In this article we report a case of NIPHS with defective cortisol response to hypoglycemia which resolved following medical treatment with diazoxide. We provide a brief overview of the existing literature, suggest potential underlying mechanisms for this complication and discuss therapeutic options and their possible mechanism of action.
CASE REPORT
A 43-year-old woman (BMI: 25.4) was admitted complaining of light-headedness, chills, lack of concentration, nervousness, muscle weakness and three recent episodes of losing consciousness followed by very low plasma glucose levels [Glu: 1.10-1.21 mmol/L (20-22 mg/dl), HbA1c: 3.7%]. Prior liver, renal, anterior pituitary and adrenal function were normal, while hormonal investigation with a supervised fast documented endogenous hyperinsulinism. Radiologic evaluation with abdominal computed tomography (CT) and magnetic resonance imaging (MRI) failed to detect an insulinoma. Treatment with diazoxide 50 mg per day had already been initiated; nevertheless, the patient did not comply with taking diazoxide, and had no proper medical follow-up. Medical and family history was unremarkable.
During her stay in our department, hypoglycemia [Glu <2.48 mmol/L (45 mg/dl)] was documented over four consecutive days following a 12-hour overnight fast. The patient had total lack of adrenergic symptoms. Other causes of hypoglycemia were excluded. Hypoglycemia was followed by inappropriately high plasma insulin (Table 1) and C-peptide levels (2.3 nmol/L) indicative of endogenous hyperinsulinism. After glucagon administration, glucose levels were increased by only 1.37 mmol/L (25 mg/dl) despite profound hyperinsulinemia [plasma insulin: 473.65 pmol/L (68.2 μIU/ml)] indicating an almost complete depletion of hepatic glycogen stores.
Cortisol and GH measurement revealed inappropriately low plasma cortisol levels [maximum: 185 nmol/L (6.7μg/dl)], appropriately increased GH levels, while ACTH, renin and aldosterone levels were normal, practically excluding the diagnosis of primary adrenal insufficiency. ACTH stimulation (Synacthen 250 μg) and corticotropin-releasing hormone stimulation (CRH) tests were performed in order to differentiate between potential secondary and tertiary adrenal insufficiency. Cortisol response was normal in both tests with a maximum of 940 nmol/L (34 μg/dl) at 60 min in the Synachten test and 726 nmol/L (26.28 μg/dl) at 45 min in the CRH test. An insulin tolerance test (ITT) to evaluate cortisol response was not performed as the patient had very low morning basal cortisol levels and complete unawareness of hypoglycemia. Due to the fact that the patient had also reported postprandial neuroglycopenia, a prolonged 3–hour 75-gr oral glucose tolerance test (OGTT) was performed. Profound hyperinsulinemia was detected after 30 minutes [glucose: 5.55 (100 mg/dl) and insulin: >1736.2 pmol/L (250 μIU/ml)], whereas asymptomatic hypoglycemia with concomitant hyperinsulinemia [Glu: 2.31 mmol/L (42 mg/dl) and insulin: 430.6 pmol/L (62 μIU/ml)] after 150 minutes.
Pancreas-focused abdominal
CT and MRI, as well as endoscopic pancreatic ultrasonography (EUS) and 111I-octreotide
scintigraphy were normal. Subsequent SACVS failed to regionalize a positive
response in a single specific region, thus allowing a therapeutic partial
pancreatectomy, since there was a positive response in all main arteries that
perfuse the pancreas.17 The SACVS procedure and the interpretation
of the results in the localization of EH are depicted in Figure 1.
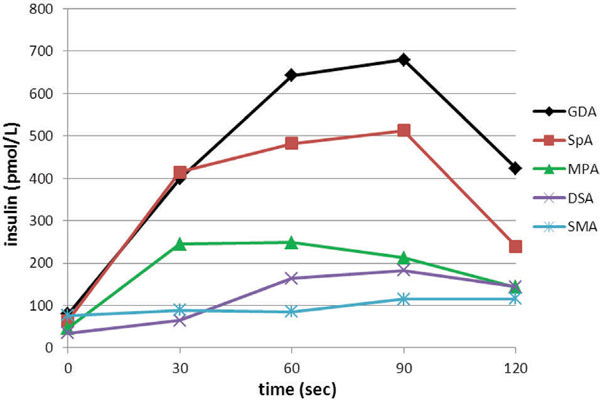
Figure 1. Insulin levels from diverse pancreatic arteries during selective arterial calcium stimulation and hepatic venous sampling (SACVS). Calcium injection into the distal part of the SpA localized increased insulin secretion to the pancreatic tail whereas calcium administration to the proximal part of SpA was indicative of increased insulin secretion in the pancreatic body. In addition, selective calcium injection into GDA, MPA and DSA which respectively supply the pancreatic head, body and tail, revealed increased insulin secretion from all the aforementioned parts of the pancreas. SACVS procedure: A catheter was placed in the right hepatic vein via femoral venous puncture. After catheterization of the femoral artery, standard pancreatic angiography was performed including selective injections of contrast agent into the gastroduodenal (GDA), proximal splenic (SpA) and superior mesenteric (SMA) artery. Following each angiogram, calcium gluconate (0.025 mEq Ca++/kg body weight) was injected into each selectively catheterized artery. In addition to the standard SACVS procedure, calcium was injected superselectively into the distal part of the SpA and into the magna pancreatic artery. The time period between each calcium injection was at least 5 minutes. Blood samples were drawn from the right hepatic vein before and 30, 60, 90 and 120 seconds after each calcium injection. A greater than 2-fold rise in insulin levels within 30-120 seconds after the injection of calcium was required to localize abnormal insulin secretion in the portion of the pancreas supplied by the artery studied. GDA: Gastroduodenal artery; SpA: Proximal splenic artery; MPA: Magna pancreatic artery; DSA: Distal part of splenic artery; SMA: Superior mesenteric artery.
Clinical data, hormonal and biochemical findings, stimulatory tests and localization studies led, by exclusion, to the diagnosis of NIPHS. Medical treatment with diazoxide (100 mg twice daily) was initiated with excellent results.
Three months after diazoxide initiation, fasting or postprandial hypoglycemias were almost absent [mean fasting and postprandial Glu: 3.83 mmol/L (69 mg/dl) and 4.33 mmol/L (78 mg/dl), respectively, HbA1c: 4.3%] while adrenergic symptoms, once absent, were reported on the two sole occasions of registered hypoglycemia [2.09 and 2.59 mmol/L (38 and 47 mg/dl)]. Six months later, although glucose levels were normal [mean fasting and postprandial Glu: 4.89 mmol/L (88 mg/dl) and 5.5 mmol/L (99 mg/dl), respectively, HbA1c: 4.8%], basal morning cortisol levels remained low [63 nmol/L (2.28 μg/dl)]. An ITT was also performed in order to validate the adrenal response and to identify if there was an increase in the glycemic threshold for cortisol hormone secretion. Cortisol and ACTH response to ITT was adequate. Nonetheless, response to ACTH, which was carried out in order to achieve restoration of normal glucose levels, was exaggerated and delayed (Table 2a). After six more months, an ITT was performed again and this time it revealed normal cortisol and ACTH response to hypoglycemia in the face of normal basal morning cortisol levels (Table 2b). ACTH and CRH stimulation tests also showed adequate cortisol response. Glucose, insulin, cortisol and GH levels before diazoxide treatment and 6 and 12 months following diazoxide treatment are shown in Table 1.
DISCUSSION
Recognition and diagnosis of rare EH causes can be challenging. We describe a case of hyperinsulinism secondary to NIPHS with impairment of cortisol response to recurrent episodes of hypoglycemia, which resolved after diazoxide treatment, and highlight the role of K-ATP channels in insulin secretion and hypoglycemia counterregulation.
NIPHS diagnosis often requires laborious biochemical and imaging investigation and can rarely be made before surgery.7 Differential diagnosis from insulinoma is important in order to evaluate the option of a possible surgical resection and can be accomplished through SACVS. In our patient, NIPHS was a diagnosis by exclusion based on biochemical findings of fasting and postprandial hyperinsulinemic hypoglycemia and positive SACVS test in diverse parts of the pancreas. Apart from the conventional arteries, further catheterization of the distal part of the splenic artery was performed in order to acquire more data on possible insulin secretion from the pancreatic body versus tail, while magna pancreatic artery was additionally catheterized as there were anatomic variations regarding other patients’ arteries (renal). In our case, ACTH levels and serum cortisol response were inappropriately low for the degree and severity of hypoglycemia, suggesting an impairment of the hypothalamic-pituitary axis. On the other hand, ACTH and CRH administration induced an adequate increase in cortisol levels confirming a centrally mediated phenomenon. ACTH response to hypoglycemia was greater in the ITT performed at 6 months following diazoxide initiation compared to that performed at 12 months, in the face of lower fasting glucose levels (3.36 vs 5.39 mmol/L). Furthermore, predominance of neuroglycopenic symptoms and lack of adrenergic ones also indicated an abnormal adrenaline response to hypoglycemia.
Counterregulatory hormones have a pivotal role in the restoration of normal serum glucose levels after hypoglycemia. Defects in counterregulation include decreased peak of hormonal (glucagon, cortisol, adrenaline) response to a hypoglycemic stimulus, abnormally decreased glycemic threshold for counterregulatory hormone release as well as hypoglycemia unawareness due to reduced sympathoadrenal responses.18-22
In patients with insulinoma, apart from reduced glucagon levels there is also a defect in GH and ACTH release in response to hypoglycemia which could be related to hyperinsulinemia-induced reduction in GHRH pulse and CRF secretion.23 Peak levels of epinephrine, norepinephrine and cortisol as well as glycemic threshold for counterregulatory hormones’ secretion were increased after insulinoma removal, suggesting that the defective hormonal response may be at least partially reversible.16 In addition, neonates with congenital hyperinsulinism have blunted serum cortisol response to hypoglycemia, whereas serum GH levels may have an appropriate response.15,24,25
K-ATP channels play a key role in glucosensing. Both, increased intracellular glucose levels of β-cells and use of sulfonylureas, inhibit efflux of potassium through the K-ATP channels, depolarizing the β-cell membrane. This opens voltage-gated calcium channels facilitating calcium influx and a rise in its intracellular levels. As a result, insulin granules fuse with the cell membrane and proinsulin is secreted.26 Components of the β-pancreatic islet glucose-sensing machinery, including K-ATP channels, have also been identified in neurons of brain glucose-sensing regions, in the hypothalamus [lateral hypothalamic area and ventromedial hypothalamus (VMH)] and areas within the hindbrain and brainstem.27-33
The exact mechanism by which low blood glucose exerts its central effects regarding defective counterregulatory hormone secretion and hypoglycemia unawareness is not known. Research focuses largely on VMH that serves as a satiety center.29-31 Experiments in rats have shown that K-ATP channels within the VMH may modulate the magnitude of counterregulatory responses to hypoglycemia by altering release of gamma-amino-butyric acid (GABA) within that region. Normally, local delivery of glucose into VMH can dose-dependently suppress glucagon and epinephrine responses to hypoglycemia through stimulation of GABA release, whereas decrease in glucose concentration may enhance the counterregulatory responses to hypoglycemia through suppression of GABA release.34 However, in recurrently hypoglycemic rats, closing of K-ATP channels in the VMH increases GABA levels thus suppressing counterregulatory responses to hypoglycemia.35 On the other hand, opening of K-ATP channels decreases GABA output, and this in turn corresponds to amplification of counterregulatory responses.36
Diazoxide reversibly inhibits glucose induced release of insulin in pancreatic beta cells by opening K-ATP sensitive channels and is used in cases of insulinoma, Dumping’s syndrome and congenital hyperinsulinism.37 Apart from a peripheral impact on β-cells, diazoxide could have a central effect in the VMH.37 It has been shown that K-ATP channels in VMH respond to diazoxide administration.38 In animal models, diazoxide caused a twofold decrease in GABA levels and increased glucagon and epinephrine responses during hypoglycemia.36
Our patient responded to diazoxide treatment not only with an elimination of hypoglycemic episodes but also with restoration of hypoglycemia awareness and of ACTH and cortisol response. Even though cathecholamine levels were not measured, recovery of adrenergic symptoms in the sole two hypoglycemic episodes that our patient had after diazoxide initiation, indicated a restoration of previously blunted adrenergic response. Diazoxide was preferred as a therapeutic option over SMS analogues due to its significantly lower cost, convenient route of administration and its potential beneficial effects on the VMH regarding glucose unawareness and restoration of counterregulatory hormones’ response.
Apart from the pivotal role of GABA on defective counterregulatory hormonal response to hypoglycemia, other substances including insulin, lactate, endogenous opioids as well as peripheral glucose sensing neurons are also implicated.39-44
In conclusion, this is the first report describing a case of NIPHS with deficient cortisol response to hypoglycemia which resolved after diazoxide treatment. Recognition, differential diagnosis and appropriate treatment of NIPHS are challenging. Mechanisms mediating defective hypoglycemia counterregulation are complex. Current research focuses on neurotransmitter signaling in diverse parts of the brain, mainly the hypothalamus. Pharmaceutical compounds, like diazoxide, that interfere with hypothalamic glucose awareness, might comprise a promising treatment option in cases of recurrent hyperinsulinemic hypoglycemia counterregulation defects.
DISCLOSURE
The authors have nothing to disclose.
REFERENCES
1. Service FJ, McMahon MM, O’Brien PC, Ballard DJ, 1991 Functioning insulinoma-incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 66: 711-719.
2. Cryer PE, Axelrod L, Grossman AB, et al, 2009 Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 94: 709-728.
3. Danforth DN Jr, Gorden P, Brennan MF, 1984 Metastatic insulin-secreting carcinoma of the pancreas: clinical course and the role of surgery. Surgery 96: 1027-1037.
4. Perren A, Wiesli P, Schmid S, et al, 2006 Pancreatic endocrine tumors are a rare manifestation of the neurofibromatosis type 1 phenotype: molecular analysis of a malignant insulinoma in a NF-1 patient. Am J Surg Pathol 30: 1047-1051.
5. Dworakowska D, Grossman AB, 2009 Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr Relat Cancer 16: 45-58.
6. Jabri AL, Bayard C, 2004 Nesidioblastosis associated with hyperinsulinemic hypoglycaemia in adults: review of the literature. Eur J Intern Med 15: 407-410.
7. Anlauf M, Wieben D, Perren A, et al, 2005 Persistent hyperinsulinemic hypoglycemia in 15 adults with diffuse nesidioblastosis: diagnostic criteria, incidence, and characterization of beta-cell changes. Am J Surg Pathol 29: 524-533.
8. Guettier JM, Kam A, Chang R, et al, 2009 Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab 94: 1074-1080.
9. Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV, 2005 Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med 353: 249-254.
10. Won JG, Tseng HS, Yang AH, et al, 2006 Clinical features and morphological characterization of 10 patients with noninsulinoma pancreatogenous hypoglycaemia syndrome (NIPHS). Clin Endocrinol (Oxf) 65: 566-578.
11. Tesfaye N, Seaquist ER, 2010 Neuroendocrine responses to hypoglycemia. Ann N Y Acad Sci 1212: 12-28.
12. Davis SN, Mann S, Briscoe VJ, Ertl AC, Tate DB, 2009 Effects of intensive therapy and antecedent hypoglycemia on counterregulatory responses to hypoglycemia in type 2 diabetes. Diabetes 58: 701-709.
13. Israelian Z, Szoke E, Woerle J, et al, 2006 Multiple defects in counterregulation of hypoglycemia in modestly advanced type 2 diabetes mellitus. Metabolism 55: 593-598.
14. Mohamed Z, Arya VB, Hussain K, 2012 Hyperinsulinaemic hypoglycaemia:genetic mechanisms, diagnosis and management. J Clin Res Pediatr Endocrinol 4: 169-181.
15. Hussain K, Hindmarsh P, Aynsley-Green A, 2003 Neonates with Symptomatic Hyperinsulinemic Hypoglycemia Generate Inappropriately Low Serum Cortisol Counterregulatory Hormonal Responses. J Clin Endocrinol Metab 88: 4342-4347.
16. Davis MR, Shamoon H, 1991 Deficient counterregulatory hormone responses during hypoglycemia in a patient with insulinoma. J Clin Endocrinol Metab 72: 788-792.
17. Doppman JL, Chang R, Fraker DL, et al, 1995 Localization of insulinomas to regions of the pancreas by intra-arterial stimulation with calcium. Ann Intern Med 123: 269-273.
18. Vella A, Service FJ, O’Brien PC, 2003 Glucose counterregulatory hormones in the 72-hour fast. Endocr Pract 9: 115-118.
19. Cryer PE, 2005 Mechanisms of hypoglycemia-associated autonomic failure and its component syndromes in diabetes. Diabetes 54: 3592-3601.
20. Siafarikas A, Johnston RJ, Bulsara MK, O’Leary P, Jones TW, Davis EA, 2012 Early loss of the glucagon response to hypoglycemia in adolescents with type 1 diabetes. Diabetes Care 35: 1757-1762.
21. Schultes B, Jauch-Chara K, Gais S, et al, 2007 Defective awakening response to nocturnal hypoglycemia in patients with type 1 diabetes mellitus. PLoS Med 4: e69.
22. Kerr D, Macdonald IA, Tattersall RB, 1989 Adaptation to mild hypoglycaemia in normal subjects despite sustained increases in counter-regulatory hormones. Diabetologia 32: 249-254.
23. Kaffel N, Chakroun E, Dammak M, et al, 2007 Paradoxal growth hormone and cortisol response to hypoglycemia caused by endogenous hyperinsulinemia: a case report. Ann Endocrinol (Paris) 68: 204-207.
24. Senniappan S, Hussain K, 2013 An Evaluation of Growth Hormone and IGF-1 Responses in Neonates with Hyperinsulinaemic Hypoglycaemia. Int J Endocrinol 2013: 638257.
25. Powell PD, Bellanné-Chantelot C, Flanagan SE, et al, 2011 In vitro recovery of ATP-sensitive potassium channels in ß-cells from patients with congenital hyperinsulinism of infancy. Diabetes 60: 1223-1228.
26. McCrimmon RJ, Evans ML, Fan X, et al, 2005 Activation of ATP-Sensitive K_ Channels in the Ventromedial Hypothalamus Amplifies Counterregulatory Hormone Responses to Hypoglycemia in Normal and Recurrently Hypoglycemic Rats. Diabetes 54: 3169-3174.
27. Routh VH, Song Z, Liu X, 2004 The role of glucosensing neurons in the detection of hypoglycemia. Diabetes Technol Ther 6: 413-421.
28. Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE, 2004 Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 53: 549-559.
29. Oomura Y, Ono T, Ooyama H, Wayner MJ, 1969 Glucose and Osmosensitive Neurones of the Rat Hypothalamus. Nature 222: 282-284.
30. Ono T, Nishino H, Fukuda M, et al, 1982 Glucoresponsive neurons in rat ventromedial hypothalamic tissue slices in vitro. Brain Res 232: 494-499.
31. Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH, 2001 Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 50: 2673-2681.
32. Balfour RH, Hansen AM, Trapp S, 2006 Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 570: 469-484.
33. Medeiros N, Dai L, Ferguson AV, 2012 Glucose-responsive neurons in the subfornical organ of the rat: a novel site for direct CNS monitoring of circulating glucose. Neuroscience 201: 157-165.
34. Zhu W, Czyzyk D, Paranjape SA, et al, 2010 Glucose prevents the fall in ventromedial hypothalamic GABA that is required for full activation of glucose counterregulatory responses during hypoglycemia. Am J Physiol Endocrinol Metab 298: E971-977.
35. Beverly JL, de Vries MG, Bouman SD, Arseneau LM, 2001 Noradrenergic and GABAergic systems in the medial hypothalamus are activated during hypoglycemia. Am J Physiol 280: R563-R569.
36. Chan O, Lawson M, Zhu W, Beverly JL, Sherwin RS, 2007 ATP-Sensitive K Channels Regulate the Release of GABA in the Ventromedial Hypothalamus During Hypoglycemia. Diabetes 56: 1120-1126.
37. Trube G, Rorsman P, Ohno-Shosaku T, 1986 Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic beta-cells. Pflugers Arch 407: 493-499.
38. Inagaki N, Gonoi T, Clement JP 4th et al 1995 Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 270: 1166-1170.
39. Spanswick D, Smith MA, Mirshamsi S, et al, 2000 Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neuroscience 3: 757-758.
40. Vele S, Milman S, Shamoon H, et al, 2011 Opioid Receptor Blockade Improves Hypoglycemia- Associated Autonomic Failure in Type 1 Diabetes Mellitus. J Clin Endocrinol Metab 96: 3424-3431.
41. Chan O, Paranjape SA, Horblitt A, Zhu W, Sherwin RS, 2013 Lactate-induced release of GABA in the ventromedial hypothalamus contributes to counterregulatory failure in recurrent hypoglycemia and diabetes. Diabetes 62: 4239-4246.
42. Watts AG, Donovan CM, 2010 Sweet talk in the brain: Glucosensing, neural networks, and hypoglycemic counterregulation. Front Neuroendocrinol31: 32-43.
43. Paranjape SA, Briski KP, 2005 Recurrent insulin-induced hypoglycemia causes site-specific patterns of habituation or amplification of CNS neuronal genomic activation. Neuroscience 130: 957- 970.
44. Reno CM, Litvin M, Clark AL, Fisher SJ, 2013 Defective counterregulation and hypoglycemia unawareness in diabetes: Mechanisms and emerging treatments. Endocrinol Metab Clin North Am 42: 15-38.
Address for correspondence:
Labrini Papanastasiou, Department of Endocrinology and Diabetes Center, Athens General Hospital “G. Gennimatas”, 154 Mesogion Avenue, 11527 Athens, Greece, Tel.: +302107768283, Fax: +302107779146, E-mail address: linapapan@yahoo.gr
Received: 04-06-2014, Accepted: 11-07-2014