1Unit of Endocrinology and Metabolic Diseases, Department of Medical and Surgical Sciences, University of Foggia, 2Radiodiagnostic Unit, “Ospedali Riuniti” University Hospital; Foggia, Italy
OBJECTIVE: Primary pigmented
nodular adrenocortical disease (PPNAD), an uncommon cause of Cushing’s
syndrome, is frequently associated with a wider clinical spectrum, the Carney
complex (CC), a multiple endocrine neoplasia syndrome.
DESIGN: We evaluated a
low-dose mitotane regimen for treating severe hypercortisolism in a 27-year-old
woman with CC. She presented with severe hypercortisolism and a history of
surgeries for breast ductal adenoma, atrial cardiac myxomas with cerebral and
peripheral arterial embolism, and near-total thyroidectomy because of an
oxyphilic adenoma. The patient refused further surgery for adrenalectomy.
RESULTS: During the first 7 months of mitotane (Lysodren, HRA Pharma, Paris,
France), the daily oral dose was progressively increased from 0.5 to 4 g/day
and then stopped because of the appearance of sustained signs of
hypoadrenalism, that required a replacement therapy with 5 mg of prednisone
o.d. A 10-month mitotane off-therapy follow-up was performed and when an
increase in urine free cortisol (UFC) was noted, the mitotane regimen was
restarted at lower doses (0.750-1 g/day). Serum morning cortisol levels and UFC
were then maintained within the normal range, with plasma mitotane ranging between
2 and 4 mg/L. A sustained regression of Cushing’s features without inducing
hypoadrenalism was achieved, which still persists after 122 months of
follow-up. Minimal initial gastric discomfort was the only side effect of which
the patient complained and only during the first higher dose mitotane course.
CONCLUSIONS: Long-term administration of a low maintenance dose of mitotane may
be suggested as treatment for hypercortisolism in CC patients who refuse or are
at high risk for surgical adrenalectomy.
Carney Complex, Cortisol, Cushing’s Syndrome, Mitotane, PPNAD
INTRODUCTION
Primary pigmented nodular adrenal dysplasia (PPNAD) is an uncommon cause of Adrenal Cushing’s Syndrome (ACS).1 PPNAD is frequently associated with a wider clinical spectrum, the Carney complex (CC), which may also include spotty skin pigmentation, heart and skin myxoma and LCCSCT (large-cell calcifying Sertoli cell tumor). CC may also be associated, although uncommonly, with acromegaly, PMS (psammomatous melanotic schwannoma), thyroid nodules or cancer, and breast ductal adenoma.2
Carney complex is an autosomal dominant multiple endocrine neoplasia syndrome due to a mutation at the 17q22–24 locus involving the tumor suppressor gene cAMP-dependent protein kinase type I-alpha regulatory subunit (PRKAR1A), coding for the type 1 regulatory subunit of PKA.3-5 PPNAD is associated with features of Cushing’s syndrome, virilization and hypertension with a lack of cortisol suppression on high dose dexamethasone (DXT) test, and paradoxical cortisol increase after 1 mg midnight DXT test in some patients,6 low plasma adrenocorticotropin hormone (ACTH) and absent cortisol and ACTH responses to corticotropin releasing hormone (CRH). As medical therapy of hypercortisolism is still far from providing effective and well-tolerated drugs, adrenal surgery remains the definitive solution to the syndrome.7 Mitotane treatment [o,p’-DDD(1-(o-chlorophenyl)-1-(p-chlorophenyl)-2,2-dichloro-ethane)] is widely used in the treatment of adrenocortical carcinoma and Cushing’s syndrome by virtue of its tissue-selective toxicity.8-11 Furthermore, mitotane may be a suitable therapeutic option for CC subjects at increased surgical risk or refusing further surgical procedures. Here we report the long-term follow up, the time-course of mitotane and the steroid serum profile of a ten-year low-dose mitotane regimen inducing correction of severe hypercortisolism in a young woman with PPNAD and CC.
PATIENT’S DESCRIPTION
A 27-year-old female CC patient had a history of surgical procedures for breast ductal adenoma, atrial cardiac myxomas with cerebral embolism and near-total thyroidectomy because of an oxyphilic adenoma. On physical examination, the patient presented with a rounded face and virilization signs and marked atrophy of the proximal muscles. No mucocutaneous manifestations of CC were observed. Laboratory findings are reported in Table 1. Molecular genetic analyses of the PRKAR1A gene revealed a common mutation: a 2-bp deletion of nucleotides T and G at positions 578 and 579. No mutations were revealed in the parents and in her sister. Nodular appearance of the left adrenal gland with typical “bead-on-a-string” PPNAD feature of the right gland was seen on computed tomography (CT) (Figure 1A). We decided to start a mitotane course before a further surgical procedure, which at the time the patient refused. During the first 7 months of the course of treatment, mitotane (Lysodren, HRA Pharma, Paris, France) was increased weekly by 0.5 g up to 4 g/day and then stopped (the cumulative dose was 602 g) because of sustained signs of hypoadrenalism (Figure 2). The mitotane regimen was restarted at lower doses (0.750-1 g/day) after a 10-month mitotane off-therapy since a gradual increase in UFC was noted. Serum cortisol levels and UFC were then maintained within the normal range, with plasma mitotane levels ranging between 2 and 4 mg/L (Figure 2). Two months from the beginning of the treatment (plasma mitotane level: 8 mg/L), a progressive regression of the typical signs of Cushing’s syndrome and hyperandrogenism with restoration of regular menses was achieved. At the end of the first mitotane treatment, the size of the adrenal glands appeared to have been reduced by the adrenolytic treatment with regression of the typical CT “bead-on-a-string” PPNAD feature of the right gland (Figure 1B), which still persists at the 122nd month (Figure 1C). Hormonal variables were measured using commercially available kits. All hormone assays were performed in the same laboratory. Plasma mitotane was determined by the HPLC method.12 Intra- and interassay coefficients of variation for all the above mentioned variables were less than 5% and 8%, respectively.
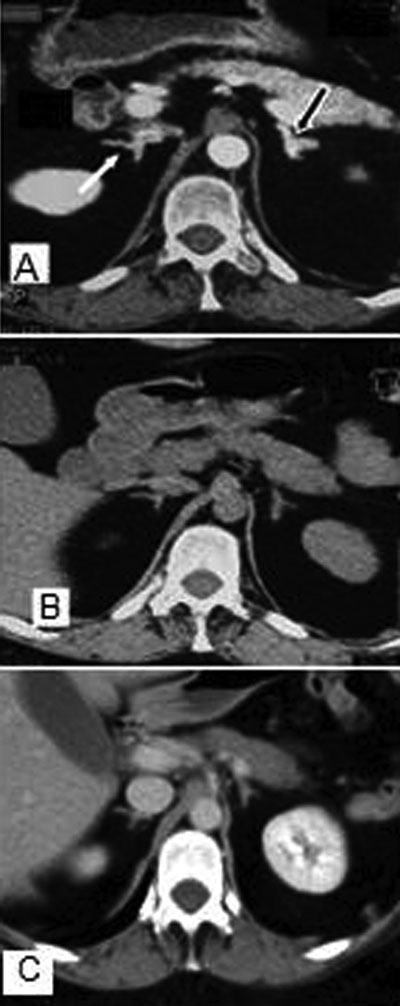
Figure 1. Computed tomography appearance of adrenal glands. (A) Enlargement of left adrenal gland (black arrow) and typical “bed on a string” feature of right adrenal gland (white arrow). Significant regression of the above characteristics after 7 (B) and 122 (C) months of MT treatment.
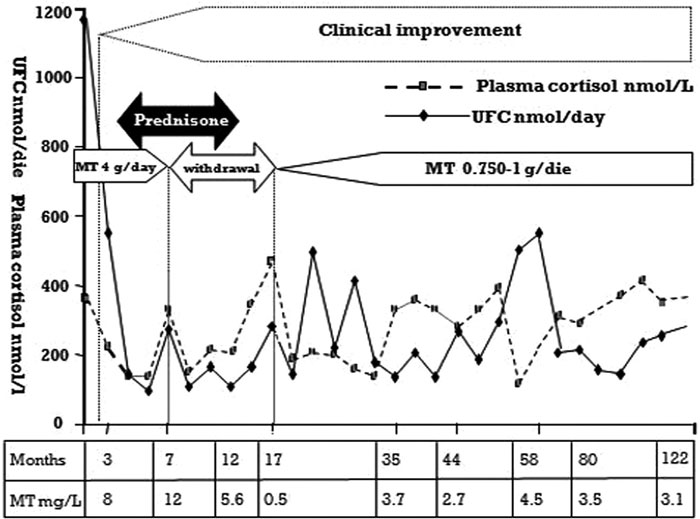
Figure 2. Serum cortisol and UFC variations and mitotane levels during the 122 months follow-up.
DISCUSSION
Due to the number of neoplastic manifestations, patients with PPNAD and CC commonly undergo several surgical procedures whose risks are aggravated by the presence of hypercortisolism or sequelae from heart myxomas and related cardiac operations. Therefore, medical therapy with mitotane may be a suitable therapeutic option for such subjects. Adrenal tissue-selective mitotane toxicity is mainly due to its local metabolic transformation of mitotane into its methylsulfonyl metabolite, MeSO2-DDE [2,2-(2-chlorophenyl,4’-chlorophenyl)1,1-dichloroethane], the human adrenal cortex also being capable of forming irreversibly bound MeSO2-[14C]DDE-protein adducts.8 Indeed, in our patient, by contrast to a pronounced reduction of plasma dehydroepiandrosterone sulfate (DEAS) and cortisol concentration, aldosterone levels were not affected by mitotane treatment. Donadille et al13 described a successful treatment of hypercortisolism in 23 ectopic ACTH secretion syndrome patients with occult and/or inoperable and/or metastatic tumors or severe hypercortisolism using long-term mitotane courses with a mean therapy duration of 1.8 ± 2.1 yr (range 0.06-9.7 y) and a mean daily dose of 3.3 ± 1.2 g. In our adult PPNAD patient, we used a well-tolerated low-dose mitotane regimen (0.750-1 g/day after initial dose of 4 g/day) and we obtained a long-term (10 years) complete and sustained regression of Cushing’s features with a significant amelioration of quality of life. Morning serum cortisol and UFC levels were maintained within the normal range with plasma mitotane ranging between 2 and 4 mg/L, a concentration much lower than that reported for adrenal carcinoma or ectopic Cushing’s syndrome patients.14 Moreover, we observed a marked reduction in DHEAS and androstenedione serum values with restoration of ovary steroids. Furthermore, during the second low-dose mitotane regimen, menses and ovary steroid plasma levels were still maintained. Surprisingly, both the circadian rhythm of serum cortisol levels and its response to 1 mg overnight DXT suppression test, which previously demonstrated a typical paradoxical increase, returned to normal levels (Table 1). These hormonal findings were associated with a considerable reduction in adrenal size (Figure 1C). Interestingly in our patient, the long-term course with low-dose mitotane did not induce hypoadrenalism. A subsequent surgical procedure for recurrence of atrial myxoma that the patient underwent was indeed well tolerated without any complications. A partial correction of bone density was also achieved. Taking into account the many surgical procedures which CC patients usually undergo, our report encourages considering low-dose mitotane treatment as a safe and effective way of achieving sustained correction of hypercortisolism by PPNAD in subjects with CC and above all for subjects at increased surgical risk or refusing further surgical procedures.
REFERENCES
1. Shenoy BV, Carpenter PC, Carney JA, 1984 Bilateral Primary pigmented nodular adrenocortical disease. Rare cause of the Cushing syndrome. Am J Surg Pathol 8: 335-344.
2. Salpea P, Stratakis CA, 2013 Carney complex and McCune Albright syndrome: An overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol 386: 85-91.
3. Stratakis CA, Jenkins RB, Pras E, et al, 1996 Cytogenetic and microsatellite alterations in tumors from patients with the syndrome of myxomas, spotted skin pigmentation, and endocrine overactivity (Carney complex). J Clin Endocrinol Metab 81: 3607-3614.
4. Horvath A, Bertherat J, Groussin L, et al, 2010 Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat 31: 369-379.
5. Stratakis CA, Kirschner LS, Carney JA, 2001 Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86: 4041-4046.
6. Louiset E Stratakis CA, Perraudin V, et al, 2009 The Paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab 94: 2406-2413.
7. Biller BM, Grossman AB, Stewart PM, et al, 2008 Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 93: 2454-2462.
8. Benecke R, Keller E, Vetter B, de Zeeuw RA, 1991 Plasma level monitoring of MT (o,p’-DDD) and its metabolite (o,p’-DDE) during long-term treatment of Cushing’s disease with low doses. Eur J Clin Pharmacol 41: 259-261.
9. Terzolo M, Daffara F, Ardito A, 2014 Management of adrenal cancer: a 2013 update. J Endocrinol Invest 37: 207-217.
10. Kerkhofs TM, Baudin E, Terzolo M, et al, 2013 Comparison of two mitotane starting dose regimens in patients with advanced adrenocortical carcinoma. J Clin Endocrinol Metab 98: 4759-4767.
11. Terzolo M, Baudin AE, Ardito A, et al, 2013 Mitotane levels predict the outcome of patients with adrenocortical carcinoma treated adjuvantly following radical resection. Eur J Endocrinol 169: 263-270.
12. Cignarelli M, Picca G, Campo M, et al, 2005 Six months mitotane course induced sustained correction of hypercortisolism in a young woman with PPNAD and Carney Complex. J Endocrinol Invest 28: 54-60.
13. Donadille B, Groussin L, Waintrop C, et al, 2010 Management of Cushing’s syndrome due to ectopic adrenocorticotropin secretion with ortho-1- para-dichloro-diphenyl- dichloro-ethane: findings in 23 patients from a single center. J Clin Endocrinol Metab 95: 537-544.
14. Baudin E, Pellegriti G, Bonnay M, et al, 2001 Impact of monitoring plasma 1,1-dichlorodiphenyldichloroethane (o,p’DDD) levels on the treatment of patients with adrenocortical carcinoma. Cancer 92: 1385-1392.
Address for correspondence:
Mauro Cignarelli, Unit of Endocrinology and Metabolic Diseases, Department of Medical and Surgical Sciences, University of Foggia, Italy; Viale Pinto, 1, 71122 Foggia, Italy; Tel.: +39 0881 732212; Fax: +39 0881 732308; E-mail: mauro.cignarelli@unifg.it
Received: 20-05-2014, Accepted: 14-07-2014