1Departments of Clinical and Experimental Medicine, University of Parma; 2Unit of Molecular Genetics, 3Departments of Paediatrics, University Hospital of Parma; 4Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Turin; Italy
OBJECTIVE: Congenital adrenal hypoplasia (CAH) is a rare disorder that can be inherited in an X-linked or autosomal recessive pattern. CAH is frequently associated with hypogonadotropic hypogonadism (HHG) with absent or arrested puberty and impaired fertility caused by abnormalities in spermatogenesis. It is estimated that more than 50% of boys with idiopathic adrenal insufficiency have mutations in the NR0B1 gene product, DAX1.
CASE REPORT: The proband is a young boy born after an uneventful pregnancy and delivery to non-consanguineous parents. At age 4 years and 4 months he came to our attention because of severe vomiting, abdominal pain, dehydration, and asthenia. The proband underwent a detailed clinical investigation including genetic testing. Sequencing analysis of the NR0B1 gene coding region from the affected child revealed a novel hemizygous deletion [c.385delC; p.(Leu129Cysfs*135)]. This mutation was also present in the heterozygous healthy mother and in her twin sister and in the first cousin of the proband. Monozygosity of the twin sisters was demonstrated. This suggests a de novo mutation and gonadal mosaicism for the deletion.
CONCLUSIONS: Adrenal hypoplasia typically presents as adrenal insufficiency during the first few months of life, however, not necessarily as shown by our index case. HHG is thought to affect all NR0B1 mutated patients who reach puberty and, as understanding of the disease has improved, more of these patients survive while presenting different features of the disease, this emphasizing the value of genetic testing in boys with primary adrenal insufficiency and suspected X-linked CAH.
Congenital Adrenal Hypoplasia, de novo mutation, Gonadal mosaicism, NR0B1 gene
INTRODUCTION
Congenital adrenal hypoplasia (CAH, OMIM #300200) is a rare disorder that can be inherited in an X-linked or autosomal recessive pattern1 and has an estimated frequency of 1:12,500 live births.2 In X-linked CAH, primary adrenocortical failure occurs because of the absence of the permanent adult adrenal cortical zone. The remaining cells, termed cytomegalic, appear larger than typical fetal adrenal cells.3,4
The clinical appearance of CAH has a bimodal phase of onset since the majority of patients present symptoms and signs within the first two months of life, but it can present also later in childhood. Adrenal insufficiency often presents with life-threatening adrenal crisis, characterized by hyperpigmentation, vomiting, poor feeding, failure to thrive, seizures, vascular collapse and sudden death. However, CAH is frequently associated with hypogonadotropic hypogonadism (HHG), a disorder that usually presents later in life, with absent or arrested puberty and impaired fertility caused by abnormalities in spermatogenesis.5
The X-linked cytomegalic form of CAH is due to mutations in the NR0B1 gene (dosage-sensitive sex reversal critical region on the X chromosome, gene 1) (OMIM#300473), which is located on the short arm of the X-chromosome (Xp21) and encodes for a 470 amino acid protein, called DAX1.1 Mutations in the NR0B1 gene were found in patients with CAH-HHG.6 This protein is a member of the nuclear hormone receptor superfamily with an unusual structure, formed of a characteristic C-terminal ligand binding domain and a novel DNA-binding domain at the N-terminus.7 DAX1 is a negative regulator that interacts with steroidogenic factor 1 (SF1, NR5A1) to inhibit SF1-mediated transactivation of numerous genes involved in the development of the hypothalamic-pituitary-adrenal/gonadal-axis and in the biosynthesis of steroid hormones.8 Furthermore, it acts as a powerful transcriptional repressor of STAR gene expression, leading to a drastic decrease in steroid production.
Since the initial identification of DAX1 as the gene responsible for CAH, several additional mutations have been discovered.9,10 All DAX1 mutations found in CAH patients alter the C-terminus of the protein, which shares similarity to the ligand-binding domain of nuclear hormone receptors and bears transcriptional repressor activity.
Here we report a child and his first cousin both with CAH carrying a novel mutation in the NR0B1 gene (c.385delC). They inherited the mutation from their respective mothers, who were monozygotic twin sisters, whereas the maternal grandparents did not present the mutation, thus providing evidence of a de novo case.
SUBJECTS AND METHODS
Clinical case
The proband is a young boy born after an uneventful pregnancy and delivery to non-consanguineous parents. No history of genetic disorders or endocrinopathies of any sort were present; however, the mother mentioned a tendency of the son to prefer salted food (salt-craving) during the 2 months before hospitalization. The family history is remarkable for hypertension in the maternal line (maternal grandfather on treatment for hypertension).
At age 4 years and 4 months, the proband came to our attention because of vomiting, abdominal pain and asthenia. He also presented with melanodermia, and hyperchromia of varicella scars.
Laboratory measurements on admission revealed severe hyponatremia (114 mmol/L), potassium at the upper level of normal (5.3 mmol/L) and hypercalcemia (5.65 mmol/L). Infusion of appropriate saline solutions initially failed to correct alterations, while loss of sodium in urine was detected leading to the diagnosis of adrenal insufficiency. High dose intravenous hydrocortisone was then started resulting in gradual improvement and normalization of serum electrolyte concentrations. Hormonal laboratory assays performed a few hours after admission revealed high basal adrenocorticotropic hormone (ACTH) level (48.84 pmol/L) and low cortisol level under this stress condition (248.4 nmol/L). Primary adrenal insufficiency was confirmed with an ACTH stimulation test (Tetracosactide 250 μg iv) (Table 1) and he was started on hydrocortisone (7.5 mg / 3 times a day, then 25 mg/m2/die in 3 doses) and fludrocortisone. During the follow-up he remained well compensated.
An autoimmune form, tuberculosis and X-linked adrenoleucodystrophy were excluded as the cause of primary adrenal insufficiency. Neurological studies were normal, as well as the abdominal ultrasound and magnetic resonance imaging (MRI).
Analysis of genomic DNA
Genomic DNA from the index case with CAH and his familial relatives (parents, grandparents and maternal aunt) was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen Inc., Valencia, CA, USA), and stored at -20°C until use. Written informed consent was obtained from all subjects included in the study and from parents for minors, as appropriate. Exons 1 and 2 of the NR0B1 gene (GeneBank accession number NM_000475.4) were amplified by PCR with intron spanning primers as previously described.11,12 Genomic DNA was sequenced using a CEQ Dye-Terminator Cycle Sequencing kit (Beckman Coulter Inc., Miami, FL, USA) according to the manufacturer’s protocol. Sequence alignments were performed with the DNAStar program (DNAStar Inc., Madison, WI, USA). Sequences with DNA variations were confirmed from a separate DNA extraction, PCR amplification and sequence reaction. All the sequence reactions were performed using a CEQ XL2000 DNA Analysis System (Beckman Coulter).
Genotype frequencies of detected polymorphisms were compared with data of the International HapMap Project (http://www.hapmap.org/), using the Utah residents with ancestry from northern and western Europe (CEU) population.
The numbering of the NR0B1 gene mutation was named according to the International recommendations for the description of sequence changes provided by the Human Genome Variation Society, HGVS (www.HGVS.org/mutnomen/). Variant description was checked using the Mutalyzer program (https://mutalyzer.nl/).13 The sequence number of the NR0B1 gene corresponding to +1 is the A of the ATG translation initiation codon. The codon number 1 is the initiation codon.
DHPLC analysis
Denaturing High Performance Liquid Chromatography (DHPLC) was performed on a Transgenomic WAVE DNA Analysis System (Transgenomic, Crewe, UK) which can detect heteroduplex formation between normal control and patient amplicons.14 One hundred and eighty-six wild-type chromosomes from healthy control subjects, having the same ethnic background as the index case, were investigated in order to exclude that the c.385delC new variant could represent a common polymorphism.
Microsatellite analysis
A set of eleven robust microsatellite markers representing different chromosomes was selected from the Mammalian Genotyping Service [http://research.marshfieldclinic.org/genetics/home/index.asp]: GTNOS, D3S3559, D3S1478, D3S2321, D1S256E, D15S994, D15S993, D15S974, D7S3036, D11S1303, D11S4114, D11S915, D14S1049 and D14S1060. These markers were chosen for their highly polymorphic index and hence have a high discriminative power to differentiate genotypes. Fluorescent amplification products were detected using a CEQ XL2000 DNA Analysis System (Beckman Coulter).
RESULTS
Sequencing analysis of the NR0B1 gene coding region and the adjacent splice sites (it contains only 2 exons) from the affected child revealed a novel hemizygous deletion of the cytosine at position 385 in the coding sequence [c.385delC; p.(Leu129Cysfs*135)], affecting amino acid number 129. Deletion would produce a frameshift mutation in the X-chromosome allele, and a premature stop codon codifying for a truncated protein of 264 amino acids, instead of 470 amino acids of the normal protein. The frameshift deletion causes the formation of 135 amino acids not belonging to the NR0B1 wild-type protein (from the affected amino acid number 129 and the premature termination codon number 264). This mutation was also present in the heterozygous healthy mother and in her twin sister, while it was absent in the maternal grandparents. This suggests ade novo mutation and gonadal mosaicism for the deletion, which is a special form of mosaicism where some gametes (oocytes in this case) carry a mutation but the rest of the cells (such as those present in peripheral blood) are normal. The cause is usually a mutation that occurs in an early stem cell that gives rise to all or part of the gonadal tissue (Figure 1) from which all or part of the gonadal tissue originates.
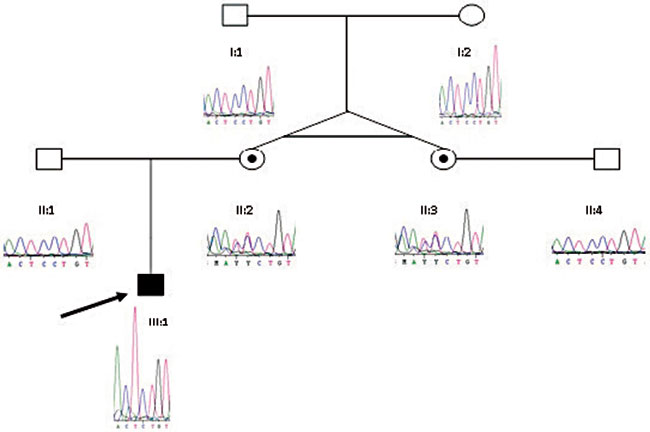
Figure 1. DNA sequence of the wild-type grandparents (I:1 and I:2), father (II:1), uncle (II:4), heterozygous mother (II:2), aunt (II:3) and affected cousin (III:2) of the hemizygous affected index case (III:1) are shown. The mutation name was c.385delC; p.(Leu129Cysfs*135) of the NR0B1 gene.
Due to the fact that the proband’s mother had a monozygotic twin sister, a prenatal test was performed also in the fetus of the proband’s aunt, who was pregnant at the time of diagnosis, and the same mutation was found.
The c.385delC mutation was absent in 186 control chromosomes, suggesting that this deletion may not represent a polymorphism.
Microsatellite analysis used to verify the monozygotic/dizygotic status of the mother and the aunt of the index case revealed that all eleven polymorphic markers were identical. Therefore, monozygosity of the twins had a probability of more than 99%. These results also confirmed the biological paternity of the two twin sisters (Table 2).
DISCUSSION
In the present work we report an CAH affected young boy with a hemizygotic NR0B1 gene frameshift mutation, which caused a loss of function allele. The child described came to our attention due to severe vomiting, abdominalgia, melanodermia, hyponatremia and asthenia typical of adrenal insufficiency at the age of 4 years and 4 months. No other clinical signs were present, his physical examination as well as his auxological parameters being within the normal range. As expected, his pubertal development was appropriate for his age and no cryptorchidism was observed.
In 1994, Muscatelli et al demonstrated that mutations in the DAX1 gene give rise to X-linked congenital adrenal hypoplasia with hypogonadotropic hypogonadism. Usually, adrenal hypoplasia presents as adrenal insufficiency during infancy, whereas hypogonadotropic hypogonadism becomes evident in affected males who survive into childhood and approach puberty. The etiology of HHG involves a combined and variable deficiency of hypothalamic GnRH secretion and/or pituitary responsiveness to GnRH resulting in low LH, FSH and testosterone.9 Animal models also indicate that DAX1 protein plays an important role in the development and function of the testis, and it has been demonstrated that increased DAX1 leads to dosage-sensitive sex reversal and a female phenotype or ambiguous genitalia in XY-genotypic males.15
The DAX1 protein, an orphan member of the nuclear receptor superfamily (NRSF) encoded by the NR0B1 gene, has the structural and functional characteristics of a transcriptional repressor7 through the LBD silencing C-terminal domain. All NR0B1 gene mutations found in CAH patients alter the C-terminus of the protein (49% of mutations are frameshift), causing the loss of DAX1 repression function. Based on these indications, the biological consequence of NR0B1 loss-of-function mutations is the overexpression of a gene, or genes, that would normally be down-regulated, leading to the failure of adrenal development and, finally, CAH associated with HHG.16 Moreover, in mice lack of Ahch, the homolog of NR0B1,17 causes progressive degeneration of the testicular germinal epithelium independent of abnormalities in gonadotropin and testosterone production and results in male sterility.
The age of presentation of CAH is typically bimodal, with the majority of patients presenting symptoms of adrenal insufficiency within the first two months of life, while in the remaining patients symptoms manifest insidiously later in childhood. This suggests a phenotypic heterogeneity but does not imply a correlation between genetic alteration and age of onset and/or severity of this condition.18 Unilateral or bilateral cryptorchidism may occur, likely because of low gonadotropin production in utero.6,19
The p.(Leu129Cysfs*135) protein change we found was described previously in a CAH affected child associated with HHG by Muscatelli et al,6 though in the latter case the stop codon was caused by a nonsense mutation (c.788T>A), while in our patient a nucleotide deletion was present. Furthermore, the familial case with CAH described by Muscatelli et al was diagnosed at 8 days of life and the HHG diagnosis was made when the patient was around 14 years. They reported that all CAH patients over 14 years and with point mutations in the NR0B1 gene were also diagnosed with HHG, confirming that this gene was responsible for both X-linked CAH and HHG, thereby providing evidence that this gene is essential for the development of a functioning hypothalamus-pituitary-gonadal axis.
In contrast to the majority of patients previously described in the literature, our patient presented onset of the disease not in the neonatal period and with absence of cryptorchidism. On account of the young age of this patient, no signs of puberty were present, as expected; however, prognostic signs for the risk of developing HHG, such as low serum gonadotropin and testosterone levels, will need to be investigated during follow-up.
The boy we have described inherited the mutation from his mother, who had a monozygotic twin sister; the proband’s maternal grandparents did not carry the mutation. The absence of the mutation in the parents of the two monozygotic twin sisters (heterozygous for the NR0B1 nucleotide deletion) may be explained by a de novo mutation and gonadal mosaicism. This is a condition where one parent’s gamete cells have the genetic defect, whereas the other cells are normal. Different samples were examined but none showed detectable mosaicism.
The proband’s cousin, diagnosed by a prenatal analysis, was born after an uneventful full-term pregnancy and carries the mutation c.385delC; p.(Leu129Cysfs*135). His physical examination revealed normal anthropometric measurements, normal male development and hyperpigmentation of the scrotum. At day 6 of life, the newborn presented blood level of glucose and sodium approaching the lower limit of normal, elevated ACTH levels, hypocholesterolemia and urinary sodium loss, and therefore he was started on hydrocortisone (2.5 mg/2 times a day), fludrocortisone (0.1 mg a day) and sodium chloride supplementation (2 mEq/ml 3 times a day). At 7 months of life, fludrocortisone was decreased (0.05 mg/day) and at the age of 1 year and 3 months he stopped sodium chloride supplementation. During the follow-up he remained well compensated.
Gonadal mosaicism for NR0B1 gene has previously been described by other authors20 in a family with CAH: the authors pointed-out the importance of genetic counselling because the existence of gonadal mosaicism indicates the possibility that the mother may have another affected male or carrier female, despite the absence of the mutation in her genomic DNA.
In conclusion, adrenal hypoplasia typically presents as adrenal insufficiency during the first few months of life. It is estimated that more than 50% of boys with idiopathic adrenal insufficiency have mutations in DAX1. On the other hand, hypogonadotropic hypogonadism is thought to affect all NR0B1 mutated patients who reach puberty,16 and, as our understanding of the disease has improved, more of these patients survive, presenting the different features of the disease. This variability of presentation and symptoms in NR0B1 gene mutated patients may reflect the contribution of other genetic factors, such as modifier genes, variability in gene expression and penetrance, and environmental factors might also be included,21 this emphasizing the value of genetic testing in boys with primary adrenal insufficiency suspected to be X-linked CAH.
Therefore, the main challenges continue to be early diagnosis and treatment of affected infants, so as to distinguish CAH from other causes of neonatal and childhood adrenal insufficiency with a view to predicting their prognosis and investigating disease associated features. This should, of course, be carried out in accordance with the therapeutic possibilities that these patients may have, including induction of puberty and long-term sex steroid replacement treatment.
FUNDING
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
AKNOWLEDGMENTS
We thank the family members for making their clinical data available.
REFERENCES
1. Weiss L, Mellinger RC, 1970 Congenital adrenal hypoplasia--an X-linked disease. J Med Genet 7: 27-32.
2. McKusick VA, Francomano CA, Antonarakis SE, 1992 Mendelian inheritance in man: Vol 1. Catalogs of autosomal dominant, autosomal recessive, and X-linked phenotypes.
3. Hay ID, Smail PJ, Forsyth CC, 1981 Familial cytomegalic adrenocortical hypoplasia: an X-linked syndrome of pubertal failure. Arch Dis Child 56: 715-721.
4. Reutens AT, Achermann JC, Ito M, et al, 1999 Clinical and functional effects of mutations in the DAX-1 gene in patients with adrenal hypoplasia congenita. J Clin Endocrinol Metab 84: 504-511.
5. Tabarin A, Achermann JC, Recan D, et al, 2000 A novel mutation in DAX1 causes delayed-onset adrenal insufficiency and incomplete hypogonadotropic hypogonadism. J Clin Invest 105: 321-328.
6. Muscatelli F, Strom TM, Walker AP, et al, 1994 Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 372: 672-676.
7. Zanaria E, Muscatelli F, Bardoni B, et al, 1994 An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature 372: 635-641.
8. Iyer AK, McCabe ER, 2004 Molecular mechanisms of DAX1 action. Mol Genet Metab 83: 60-73.
9. Jadhav U, Harris RM, Jameson JL, 2011 Hypogonadotropic hypogonadism in subjects with DAX1 mutations. Mol Cell Endocrinol 346: 65-73.
10. Wheeler B, George PM, Mackenzie K, Hunt P, Potter HC, Florkowski CM, 2008 Three cases of congenital adrenal hypoplasia with novel mutations in the (NROB1) DAX-1 gene. Ann Clin Biochem 45: 606-609.
11. Achermann JC, Ito M, Silverman BL, et al, 2001 Missense mutations cluster within the carboxyl-terminal region of DAX-1 and impair transcriptional repression. J Clin Endocrinol Metab 86: 3171-3175.
12. Ostermann S, Salvi R, Lang-Muritano M, et al, 2006 Importance of genetic diagnosis of DAX-1 deficiency: example from a large, multigenerational family. Horm Res 65: 163-168.
13. Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE, 2008 Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat 29: 6-13.
14. Xiao W, Oefner PJ, 2001 Denaturing high-performance liquid chromatography: A review. Hum Mutat 17: 439-474.
15. McCabe ER, 1996 Sex and the single DAX1: too little is bad, but can we have too much? J Clin Invest 98: 881-882.
16. Phelan JK, McCabe ER, 2001 Mutations in NR0B1 (DAX1) and NR5A1 (SF1) responsible for adrenal hypoplasia congenita. Hum Mutat 18: 472-487.
17. Guo W, Lovell RS, Zhang YH, et al, 1996 Ahch, the mouse homologue of DAX1: cloning, characterization and synteny with GyK, the glycerol kinase locus. Gene 178: 31-34.
18. Verrijn Stuart AA, Ozisik G, de Vroede MA, et al, 2007 An amino-terminal DAX1 (NROB1) missense mutation associated with isolated mineralocorticoid deficiency. J Clin Endocrinol Metab 92: 755-761.
19. Peter M, Viemann M, Partsch CJ, Sippell WG, 1998 Congenital adrenal hypoplasia: clinical spectrum, experience with hormonal diagnosis, and report on new point mutations of the DAX-1 gene. J Clin Endocrinol Metab 83: 2666-2674.
20. Zhang YH, Guo W, Wagner RL, et al, 1998 DAX1 mutations map to putative structural domains in a deduced three-dimensional model. Am J Hum Genet 62: 855-864.
21. McCabe ER, 2007 DAX1: Increasing complexity in the roles of this novel nuclear receptor. Mol Cell Endocrinol 265-266: 179-182.
Address for correspondence:
Davide Martorana, PhD, Unit of Molecular Genetics, University Hospital of Parma, Via Gramsci, 14, 43126 Parma, Italy, Tel.: +390521033059, Fax: +390521293294, Ε-mail: dmartorana@ao.pr.it
Received 12-03-2014, Accepted 14-04-2014