1Endocrine Research center, Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Sciences; 2Department of Surgery, Taleghani General Hospital, Shahid Beheshti University of Medical Sciences; 3Department of Pathology, Taleghani General Hospital, Shahid Beheshti University of Medical Sciences; 4Department of pathology, Rasool Akram medical complex, Iran University of Medical Sciences; Tehran, Iran; 5Department of Clinical Genetics, Churchill Hospital; 6Oxford Centre for Diabetes, Endocrinology and Metabolism, Churchill Hospital, University of Oxford; Oxford, UK
Succinate Dehydrogenase-B (SDH-B) gene mutations constitute one of the most frequent forms of hereditary paragangliomas (PGL). Genetic study is advised in all cases for the evaluation of tumour behaviour, the selection of optimal management and the surveillance of the first degree relatives. There are limited data on the genetic characteristics of patients with PGLs from Middle East countries, and to our knowledge this is the first study from Iran. We present the clinical and genetic characteristics of a 29-year old woman who presented with hypertension secondary to a para-aortic PGL. She was shown to have a novel mutation in the SDH-B gene and her family was subsequently screened. We also emphasize the problems in diagnosing and treating patients in this region.
Family, Iran, Paraganglioma, Phaeochromocytoma, SDH-B
INTRODUCTION
Phaeochromocytomas (PCC) and paragangliomas (PGL) are neuroendocrine tumours arising from chromaffin cells of the adrenal medulla or extra-adrenal sympathetic or parasympathetic ganglia, respectively. The incidence of these tumours is approximately 2-8 per million population per year, with extra-adrenal tumours comprising some 10-20% of the cases. Mostly, the term phaeochromocytoma is used for all catecholamine-secreting tumours located either in adrenal or in extra-adrenal locations, while the term paraganglioma is used only for head-and-neck tumours originating from the parasympathetic system. Several authors, however, restrict the term PCC to specifically adrenal tumours and label all others as extra-adrenal PGLs.1,2 Most PGLs are located in the abdominal or pelvic region and may present with severe hypertension due to the secretion of large amounts of adrenaline or noradrenaline from the tumour. PGLs develop as sporadic or hereditary forms, but germline mutations are now considered to be responsible for some 30-40% of patients presenting with phaeochromocytomas and 50% of those with paragangliomas. Mutation in the succinate dehydrogenase-B gene (SDH-B), which encodes for subunit B of the complex enzyme, has been shown to be one of the most prevalent forms of the hereditary variants of PGL, with a high rate of malignant transformation.3,4 There are only very limited reports on hereditary forms of PGLs in countries of the Middle East, and this is the first study addressing the clinical and genetic characteristics of an Iranian family harbouring a novel SDH-B mutation.
CASE PRESENTATION
A 29-year old woman was referred to Taleghani General Hospital, Tehran, Iran, in 2011 because of persistent headache and palpitations for the previous 6 years which had recently deteriorated. Her past history was positive for a single episode of severe arterial hypertension (blood pressure 270/110mmHg) that was incidentally discovered during maxillo-facial surgery 5 years previously. The surgery was cancelled and evaluation of the patient revealed high 24h urinary metanephrine (800µg/24h, normal <440µg/day) and 24h VMA (17mg/day, normal <12mg/day) excretion, but with reported normal abdominal MRI and CT scans. The patient refused further evaluation and was discharged on combined adrenoceptor blockade with prazosin and atenolol. Five months prior to admission she was seen at another hospital for increasing headache and palpitations: an abdominal CT scan showed a para-aortic tumour. She underwent a percutaneous needle biopsy and the mass was diagnosed as a paraganglioma. She was referred to our hospital for further evaluation.
On physical examination her blood pressure was 170/110mmHg and heart rate was 110 beats per minute: the rest of the physical examination was negative. Routine laboratory evaluations were normal except for mild normocytic anaemia. Laboratory evaluation revealed high 24h urine VMA (20.3mg/day, normal <12) and normetanephrine (3000µg/day, normal <440). Abdominal CT and an MRI showed a lobulated 3cm round mass in the para-aortic region (Figure 1, black arrows).

Figure 1. Abdominal CT scan showing the right sided para aortic tumour.
After appropriate management of her hypertension with modification to her α- and β-adrenoceptor blockade, she underwent surgery and a solitary 4 by 3.5 by 2.5 cm mass located in the para-aortic region with an associated enlarged lymph node were removed. Histopathologic evaluation revealed a well-encapsulated tumour with a reddish-brown fleshy cut surface. The cells were bound with a delicate highly vascular fibrous stroma and arranged in well-defined nests or zellballen pattern. The tumoural cells had finely granular cytoplasm, round nuclei and prominent nucleoli (Figure 2A). Immunohistochemistry evaluation revealed positive staining for chromogranin A in the tumour cells, positive S100 in the sustentacular cells, a Ki-67 of 1% and negative staining for cytokeratin in the tumour cells (Figure 2B). The lymph node was free of tumour. The histopathologic diagnosis was of a paraganglioma.

Figure 2A. Characteristic nesting pattern of tumour cells (H&E×100). 
Figure 2B. Nests of tumoural cells strongly immunoreactive to chromogranin A (immunoperoxidase stain for chromogranin A×100).
Genetic analysis in Oxford on DNA samples prepared from the peripheral white cells revealed a heterozygous 3-nucleotide deletion, c.596-598 delACT (p.Tyr199del) in exon 6.
The patient’s post-operative course was uneventful. Her blood pressure normalised after surgery and the patient was discharged from hospital after 5 days. On her last examination, one year after surgery, she had mild diastolic hypertension (130/100 mmHg) on atenolol 50 mg and hydrochlorothiazide 25 mg/day without any signs or symptoms of catecholamine excess, and her urinary metanephrine excretion had normalised.
The patient was the offspring of a consanguineous marriage, her parents being cousins on the maternal side. First-degree family members including her father, mother, three brothers and three sisters were also assessed. Both parents were hypertensive, 160/90 mmHg and 170/120 mmHg, but had no symptoms suggestive of catecholamine excess; no other family member agreed to clinical examination and none to biochemical assessment. However, all agreed to genetic analysis and the same mutation was found in the mother and all the siblings except one of the sisters (Figure 3).
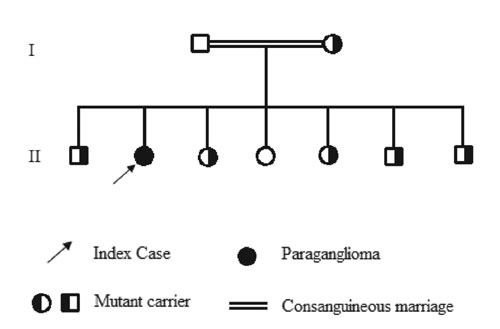
Figure 3. Pedigree of the family.
DISCUSSION
Extra-adrenal tumours comprise 10 to 20% of cases of PCC/PGLs and are more likely to be malignant in comparison to intra-adrenal tumours, 20-50% versus 10-15%. Recent figures suggest that some 40-50% of patients with PGLs suffer from a hereditary form of the disease, but this will depend on the numbers of genes and also the ethnic and geographic backgrounds of the population studied.5-7 Currently, there are known to be hereditary forms of the disease that are due to mutations of principally 11 genes: NF1, VHL, RET, SDH (A, B, C, D), SDHAF2, TMEM127, HIF2A and MAX.8-10
Succinate Dehydrogenase (SDH) is a tetrameric protein that is attached to the inner mitochondrial membrane and has 4 subunits (A, B, C and D) which are necessary for the functional activity of the protein. The enzyme catalyses succinate to fumarate in the Krebs cycle and contributes to electron transport; its main function is to link the products of the Krebs cycle to the electron transport system in order to complete glucose metabolism and ATP synthesis. The SDH-B gene is located on chromosome 1, spans 35.4 Kb, contains 8 exons and encodes for a 280 amino-acid protein.7
Mutations in the SDH-B gene are associated with the PGL4 syndrome. While the precise mechanism of tumorigenesis in carriers of the SDH-B mutation is unclear, it seems that increases in succinate lead to activation of hypoxia-inducible factors (HIF) due to a block of their ubiquitination: this then leads to deregulation of target genes implicated in cell proliferation, apoptosis and angiogenesis that finally results in malignant transformation.11-13 SDH-B gene mutation as the pathogenetic mechanism in the development of hereditary PGLs was originally described by Astuti et al in 2001;14 the following year, 2002, Neumann et al demonstrated that 66 out of 271 (24%) of patients from Germany and Poland who were classified as sporadic and non-syndromic PCC/PGL indeed had hereditary forms of the disease and had a mutation in one of their corresponding genes. According to their study, SDH-B mutation positive carriers comprised 5.4% of the patients.3 Subsequently, Manelli et al showed that 32.1% of their group of 501 patients had a hereditary form of the disease. They also showed that prevalence of the hereditary form varies from 11% to 100% in different subgroups according to clinical characteristics of patients such as age, location of the tumour, the number of tumours, familial clustering or association of syndromic manifestation. SDH-B mutation carriers comprised 4.8% of their patients.4 The study conducted by a French group on 445 patients with extra-adrenal PGLs demonstrated that 54% of their patients were SDH mutation carriers, those with SDH-B mutation comprising 22% of the cases.15
More than 90% of mutations are either a point mutation or small deletions or insertions in exons 1-7. Large deletions are found in less than 10% of the cases. No mutation has yet been found in exon 8. There does not appear to be a clear genotype-phenotype correlation.2
SDH-B mutation carriers may present with either thoraco-abdomino-pelvic (TAP) or head-and-neck tumours (HNPGLs). The most frequent form is a solitary, catecholamine-secreting TAP tumour. The median age at diagnosis is 34 years, but children less than 10 years may also be affected. Of SDH-B mutation carriers, 29% develop the tumour by age 30 years and 45% by age 40 years;16 10% of cases are hormonally silent, which may be due an absence of tyrosine hydroxylase, the rate-limiting enzyme of catecholamine synthesis.17 According to the study by Ricketts et al on 295 patients with SDH-B mutation, the risks of TAP and HNPGL tumour development were 52% and 29%, respectively, by age 60 years.18
One specific feature of SDH-B mutation carriers is their tendency to develop a malignant form of PGL, while they may also develop tumours of other organs such as renal cell carcinoma19,21 or gastrointestinal stromal tumours.20 Neumann et al reported that 34% of their patients with an SDH-B mutation had malignant PGLs; they also reported two cases of renal cell carcinoma and one case of papillary thyroid carcinoma among their patients.21
Genetic study is essential for risk stratification, selection of therapeutic modalities, assessment of tumour behaviour and also the evaluation of first-degree relatives.2,22 Currently, most large facilities specialising in the diagnosis and treatment of these patients will undertake multiple gene screening, although where facilities are limited the clinical characteristics of the patients such as age, family history, presence or absence of hypertension, localisation and solitary or multiple nature of the tumour can be helpful in the prioritisation of genetic screening.1,15
With regard to the current family, the mutation c.596-598 Del ACT (p.Tyr199del) is novel and has not previously been reported. As shown in Figure 3, the mother is involved, as are all of the siblings other than one sister. Evaluation of the family members such as collection of urine and imaging could not be completed because they lived in a remote area and were not willing to pursue the study, so we cannot be certain if there have been other cases with tumour among the family members, especially since two of them were hypertensive. This demonstrates the problems in screening and identifying patients with genetic disorders in societies where the economy is limited and specialists, clinicians and infrastructure are of limited availability.
The p.Tyr199 resides within a highly conserved iron-sulphur binding domain (4Fe-4S), which plays a crucial role in the mitochondrial electron transport system. Residue 199 is highly conserved across species and is in a highly conserved domain of the protein. Recently, an in-frame deletion of a different amino-acid in this domain, p.Ser195del, has been reported in a patient with a PGL and in his affected son.23 Based on these data, we believe that this mutation is highly likely to be pathogenic. With respect to the young age of the family members and low penetrance rate of SDH-B gene mutation, which is about 25-40%, prolonged follow-up of the patient and indeed the family is needed for accurate interpretation.24 In general, in mutation-positive subjects, regular measurement of plasma and/or urinary metanephrines, at 6-monthly or yearly intervals, is the recommended follow-up plus annual MRI of neck, thorax and abdomen and radionuclide scanning. Radilobelled 123I-MIBG is relevant therapeutically as patients can be selected for 131I-MIBG therapy, but the sensitivity is not high, especially for metastatic tumours.25 FDG-PET is preferable for sensitive detection because, as the biochemical derangements render these tumours highly inefficient in their use of glucose, they thus show a high degree of glucose uptake. Other radionuclides can be used for PET scanning but are of very limited availability.25
In conclusion, in this paper we present for the first time the clinical and genetic characteristics of a family with a novel SDH-B mutation from Iran.
ACKNOWLEDGMENTS
The authors wish to thank the family members for their cooperation in the study.
FUNDING
The study was financially supported by the Research Institute for Endocrine Sciences (RIES) Shahid Beheshti University of Medical Sciences, Tehran, Iran.
REFERENCES
1. Neumann HP, Eng C, 2009 The approach to the patient with paraganglioma. J Clin Endocrinol Metab 94: 2677-2683.
2. Fishbein L, Orlowski R, Cohen D, 2013 Pheochromocytoma/Paraganglioma: Review of perioperative management of blood pressure and update on genetic mutations associated with pheochromocytoma. J Clin Hypertens (Greenwich) 15: 428-434.
3. Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group, 2002 Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346: 1459-1466.
4. Manelli M, Castellano M, Schiavi F, et al; Italian Pheochromocytoma/Paraganglioma network, 2009 Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 94: 1541-1547.
5. Fishbein L, Nathanson KL, 2002 Pheochromocytoma and paraganglioma: understanding the complexities of the genetic background. Cancer Genet 205: 1-11.
6. Muth A, Abel F, Jansson S, Nilsson O, Ahlman H, Wängberg B, 2012 Prevalence of germline mutations in patients with pheochromocytoma or abdominal paraganglioma and sporadic presentation: a population-based study in Western Sweden. World J Surg 36: 1389-1394.
7. Pasini B, Stratakis CA, 2009 SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 266: 19-42.
8. Jiang S, Dahia PL, 2011 Mini review: the busy road to pheochromocytomas and paragangliomas has a new member, TMEM127. Endocrinology 152: 2133-2140.
9. Fishbein L, Merrill S, Fraker DL, et al, 2013 Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 20: 1444-1450.
10. Zhuang Z, Yang C, Lorenzo F, et al, 2012 Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 367: 922-930.
11. Pollard PJ, El-Bahrawy M, Poulsom R, et al, 2006 Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab 91: 4593-4598.
12. Favier J, Gimenez-Roqueplo AP, 2010 Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab 24: 957-968.
13. López-Jiménez E, Gómez-López G, Leandro-García LJ, et al, 2010 Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol 24: 2382-2391.
14. Astuti D, Latif F, Dallol A, et al, 2001 Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69: 49-54.
15. Burnichon N, Rohmer V, Amar L, et al, 2009 The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab 94: 281-287.
16. Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al, 2005 Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 91: 827-836.
17. Timmers HJ, Kozupa A, Eisenhofer G, et al, 2007 Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 92: 779-786.
18. Ricketts CJ, Forman JR, Rattenberry E, et al, 2010 Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 31: 41-51.
19. Vanharanta S, Buchta M, McWhinney SR, et al, 2004 Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 741: 153-159.
20. Pasini B, McWhinney SR, Bei T, et al, 2008 Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 16: 79-88.
21. Neumann HP, Pawlu C, Peczkowska M, et al; 2004 European-American Paraganglioma Study Group. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292: 943-951.
22. Erlic Z, Rybicki L, Peczkowska M, et al, 2009 Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res 15: 6378-6385.
23. Mhatre AN, Li Y, Feng L, Gasperin A, Lalwani AK, 2004 SDHB, SDHC, and SDHD mutation screen in sporadic and familial head and neck paragangliomas. Clin Genet 66: 461-466.
24. Jafri M, Maher ER, 2012 The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol 166: 151-158.
25. Timmers HJ, Chen CC, Carrasquillo JA, et al, 2009 Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 94: 4757-4767.
Address for correspondence:
Prof. Ashley Grossman, OCDEM, University of Oxford, OX3 7LE, UK, E-mail: ashley.grossman@ocdem.ox.ac.uk
Received: 09-10-2013, Accepted: 06-11-2013