Department of Endocrinology, Internal Medicine and Clinical Chemistry, Max-Planck-Institute of Psychiatry; Munich; Germany
BACKGROUND: Leydig cell hypoplasia is a rare autosomal recessive condition that interferes with the normal development of male external genitalia in 46,XY individuals. It is mediated by mutations in the lutropin/choriogonadotropin receptor gene, resulting in the impairment of either the binding of hormone or signal transduction.
OBJECTIVE/DESIGN: We report a 32-year-old female patient with severe Leydig cell hypoplasia due to a novel homozygote nonsense mutation in exon 10 (c.907C>T, p.Gln303Ter) of the lutropin/choriogonadotropin receptor gene. Interestingly, a second mutation was found (c.935A>G, p.Asn312Ser) downstream of the disruption of the gene sequence.
CONCLUSIONS: This case report demonstrates the coexistence of a novel homozygote nonsense mutation with a second mutation in the same hormone binding domain, expanding the genotypic spectrum of lutropin-choriogonadotropic hormone receptor gene mutations. The first diagnosis of this mutation in an adult 46,XY female patient from Morocco underlines the importance of thorough clinical and genetic examination, not only in pre- and post-pubertal children but also in adults originating from conservative socio-cultural backgrounds.
Disorder of sexual development, DSD, Leydig cell hypoplasia, LHCGR mutation
INTRODUCTION
Leydig cell hypoplasia (LCH) is a rare autosomal recessive condition that interferes with the normal development of male external genitalia in 46,XY individuals. It is mediated by mutations in the lutropin-choriogonadotropic hormone receptor (LHCGR) gene. The most common cause for functional impairments is decreased or absent receptors on the cell surface due to misfolding of the receptors and their retention in the ER,1-3 while another result is the impairment of either luteinizing hormone (LH)/human chorionic gonadotropin (hCG) binding or signal transduction.4
The LHCGR belongs to the large family of G-protein-coupled receptors (GPCRs). The receptor gene, located on chromosome 2p21, consists of 11 exons and 10 introns, resulting in an overall protein length of 674 amino acids.5 The common structure is characterized by a typical seven-transmembrane signal transduction domain (7TDM) which is essential for the signal transduction and activation of the heterotrimeric G proteins.4 The LHCGR plays an essential role in sex development and reproduction. During embryologic and foetal development, it is predominantly activated by the hCG secreted by the placenta. This activation initiates Leydig cell proliferation and subsequently induces foetal testosterone production. To date, more than 50 different mutations in this particular gene have been described, which lead to a wide range of phenotypic alterations.6,7 A basic dichotomous classification can be made by dividing these mutations into activating or inactivating mutations.
Inactivating mutations of the LHCGR gene, depending on their location and extent, may result in varying degrees of LCH. In the extreme, they may affect male sex differentiation in such a way that foetal testosterone concentrations are too low to stabilize the mesonephric duct and therefore may lead to a 46,XY disorder of sexual development (DSD) (Type 1 of LCH). However, as the function of the Sertoli cells is usually unaffected by these mutations, the secreted anti-Mullerian hormone (AMH) leads to the degradation of Mullerian structures, while the external genitalia are female.8 Since testosterone is crucial for the descent of the testicles, the clinical picture also includes cryptorchidism. Less severe forms of LCH may only result in a mild undervirilization with the presence of a micropenis or hypospadias (Type 2 of LCH).3 Since LH is non-essential for early female development, 46,XX children with these mutations may first show clinical abnormalities at the age of puberty. These girls therefore present with primary amenorrhoea and might have to depend on life-long sex-steroid replacement therapy.9 In the present study, we report the case of a 32-year-old female patient with severe Leydig cell hypoplasia due to a novel homozygote nonsense mutation in the LHCGR gene that expands the genotypic spectrum of LHCGR mutations.
AIMS AND METHODS
Case report
We report the case of a 32-year-old phenotypically female patient from Morocco who was referred to our department due to incomplete pubertal development and primary amenorrhoea. A physical examination revealed infantile breast development and modest pubic hair growth (B2 and PH4, respectively). The subsequent gynaecological examination demonstrated the presence of a short hypoplastic vagina of 3 cm in length. Based on the assumption of the presence of a 46,XY disorder of sexual development, we then performed a karyotype analysis which supplied evidence of an inconspicuous 46,XY karyotype. The consecutively performed magnetic resonance imaging (MRI) of the abdomen led to the detection of bilateral inguinal gonads, with an overall volume of 10ml, which were morphologically identified as testicles. As suspected, the imaging results also confirmed the absence of any Mullerian structures such as a uterus or fallopian tubes. The patient had negative family history regarding DSD. At first presentation, serum LH was about 3 times the upper normal limit (33 mU/ml, reference range 0.8–8.3 mU/ml), while testosterone was in the female adult normal range (0.7 nmol/l). Estradiol was significantly low (10.6 pg/ml) and serum FSH (7.7 mU/ml, reference range 2-10 mU/ml) was unremarkable. AMH (67.5 µg/l, reference range 1.5-4.3 ug/l) and inhibin b (548 ng/l, reference range 130-400 ng/l) were both above the upper limit, underlining the presence of functioning Sertoli cells. Dehydroepiandrosterone sulphate (DHEAS), 17-hydroxyprogesterone (17OHP) and sex hormone-binding globulin (SHBG) were unremarkable, as was dihydrotestosterone (DHT). According to the laboratory results and the clinical presentation, there was no indication of an androgen receptor defect or of 5-alpha-reductase or 17-alpha-hydroxysteroid dehydrogenase deficiencies, observations that were also excluded with molecular analysis. Since all the findings were compatible with LCH, we performed a genetic analysis focusing on an inactivating mutation in the LHCGR gene.
Genetic analysis of the LHCGR gene
After obtaining informed consent of the patient, genomic DNA was extracted from peripheral blood. Exons 1-11 and adjacent exon-intron boundaries were amplified by polymerase chain reaction (PCR). The PCR fragments were sequenced after purification from the agarose gels. Sequencing of the LHCGR gene on chromosome 2p21 revealed a novel homozygote mutation p.Gln303Ter (c.907C>T) in exon number 10 of the LHCGR gene, causing the replacement of cytosine by thymine at coding position 907, which was followed by the formation of a premature stop codon sequence (Ter). The mutation was located in the C-terminal cysteine-rich cluster region (CCR), also referred to as the “hinge region” (HR), of the LHCGR. Consequently, protein synthesis is stopped at this position, leading to the formation of a truncated protein. Interestingly, we also detected a second mutation, p.Asn312Ser (c.935A>G), downstream of the first mutation (Figure 1).
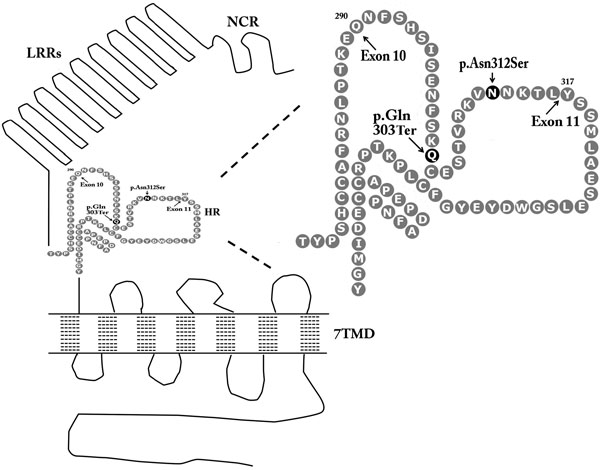
Figure 1. Graphical arrangement of the luteinizing hormone/choriogonadotropin receptor (LHCGR) of this patient. LRRs: Leucine-rich repeats, 7TDM: Seven-transmembrane signal transduction domain, NCR: N-terminal cysteine-rich cluster, HR: Hinge region, Gln: Glutamine, Ter: Termination Codon, Asn: Asparagine, Ser: Serine.
Unfortunately, due to the patient’s immigrant background, we could not perform genetic analysis of the parents to characterize the novel mutation as germline or de novo, but further enquiry confirmed the consanguinity between the parents (cousins), leading to the hypothesis that the heterozygous mutation would probably have been present in them as well. The reasons for the delay in seeking medical help may have been socio-cultural factors. After psychiatric counselling, confirming a normal female gender identity, vaginoplastic surgery was performed as well as a bilateral orchidectomy due to the malignancy risk. The histopathological evaluation of the testis revealed fibrosis. The patient was started on a transdermal sex steroid substitution with estradiol.
DISCUSSION
In the present study we report a novel homozygote nonsense mutation (p.Gln303Ter) accompanied by a second downstream mutation (p.Asn312Ser, (c.935A>G)) in the LHCGR gene of a phenotypically female patient leading to 46,XY DSD. The first mutation has not previously been described up to now according to the “The Human Gene Mutation Database” (HMGD), the Institute of Medical Genetics in Cardiff (http://www.hgmd.cf.ac.uk) and Medline search (http://www.ncbi.nlm.nih.gov/pubmed) and appears to be novel, whereas the second mutation has been described, and has also been characterized as an activating mutation leading to gonadotropin-independent precocious puberty in a short clinical report by Mitre and Lteif in 2009.10
For a better understanding of the effects induced by the different mutation sites, it is necessary to take a closer look at the molecular receptor-architecture (Figure 1). As mentioned before, the LCHGR composes a 7TMD which is connected to an N-terminal extracellular ligand-binding domain (ECD).11 All glycoprotein hormone receptors comprise a series of leucine rich repeats (LRRs) in their ECD, which are flanked at each end by N-terminal and C-terminal cysteine-rich clusters (NCR, CCR).12
The nonsense mutation found in our case subject is located at coding position 907 of exon 10 (Figure 1). This particular region constitutes an essential part of the extracellular domain of the LHCGR, since it is situated at a signal specific subdomain, the so-called hinge region (HR), of the receptor. Though not directly involved in the ligand-binding process, the HR is an essential contributor to receptor functioning. This region, which is situated near the C-terminal end of the extracellular domain, contains two sequence motifs which are coupled by disulfide bonds, thought to be essential for structural stabilization.13
The exact topology of the HR of the LHCGR has not been completely elucidated so far.14 However, there is structural information available on the TSH-receptor and just recently the crystal structure of the entire extracellular domain of the follicle-stimulating hormone receptor (FSHR) has been described.13 Since these glycoprotein hormone receptors share great homology, it allows speculation on the structure and function of this particular region of the LHCGR. In contrast to earlier reports, indicating that the HR forms a separate structural unit with high structural flexibility, Jiang et al just recently demonstrated that it should rather be regarded an integrated part of the ectodomain which is not amenable to large-scale conformational changes. In detail, the FSH-receptor constitutes a sulfotyrosine (sTyR) site in the hinge region which facilitates signal transduction in such a way that the hormone binds to the high-affinity hormone-binding subdomain and this reshapes the ligand conformation to form a sTyr-binding pocket. Interestingly, the FSHR subsequently inserts its sTyr into the FSH nascent pocket, which eventually leads to receptor activation and signal transduction. A similar mechanism is suggested for the sulfated tyrosine Y331 of LHCGR by site-directed mutagenesis.15 This underlines the importance of the hinge region for receptor functioning so that any affecting mutation may have a fundamental impact on ligand-receptor interaction and subsequent signal transduction.
Different inactivating nonsense mutations have been found in regions of the extracellular domain.16 The loss of function seen in these mutations is caused by the impaired protein transport from the endoplasmic reticulum to the plasma cell membrane and dysfunctional protein folding.
In our case, the identified nonsense mutation, leading to the formation of a stop codon, resulted in the complete termination of the protein translation process and hence to the translation of a protein remnant lacking large parts of the HR and the entire 7TMD. We did not study in vitro the mutation identified in our patient, as it was quite obvious that the complete lack of the 7TDM would result in a truncated receptor which cannot be expressed at the cell surface. Furthermore, even if the protein was stably expressed, it would not have signalling capacities due to the importance of the serpentine domain for G-protein coupling and signal transduction. In addition, following selective deletion studies in vitro, it has been demonstrated that complete deletion of the hinge region alone leads to total unresponsiveness of the investigated cells for LH and hCG.15 This is, independent of the 7TDM loss, solely explained by the presumed inability of the receptor remnant to be expressed at the cell surface.12 It has been shown that, the replacement of the intercysteinic stretches of amino acids 305-335, which are essential for disulfide bounding and therefore stability of the HR lead to a sharp decrease of LH potency.
Looking at the second mutation found in our patient in amino acid 312, it can be speculated that this homozygote substitution of adenine by guanine has not led to substantial change in receptor functioning and signalling, as was the case with the first homozygote mutation in amino acid 303 which led to a truncated protein. However, it has once been reported that this mutation of the extracellular domain could be potentially an activating mutation leading to testotoxicosis. Mitre and Lteif described the case of a young boy who was heterozygous for the p.Asn312Ser mutation and suffered from gonadotropin-independent precocious puberty.12 It should be emphasized though that this is the only case in the available literature that reports an activating mutation in a region other than the transmembrane domain and exon 11, as the sixth transmembrane domain, the third intracellular loop and recently the seventh transmembrane domain seem to be the mutational hot spots of these alterations.16,17 The finding of an activating mutation in the extracellular domain of the TSH receptor indicates, however, that similar activating mutations in the LHCGR cannot be excluded. In our case, the effect of the second mutation was attenuated due to the coexistence with the first nonsense mutation and no causal relationship with the phenotype can be proven.
Another interesting aspect from the clinical point of view is the delay in seeking medical advice, despite the long course of the primary amenorrhoea, a fact that could be attributable to the socio-cultural background of the patient. Though the patient had been married for several years, we could not obtain any further information regarding sexual intercourse before the vaginoplastic surgery, as the patient declined to answer these intimate questions.
CONCLUSIONS
In summary, we describe a phenotypically female patient with 46,XY DSD with a novel homozygote nonsense mutation (p.Gln303Ter) accompanied by a second homozygote mutation (p.Asn312Ser) in the LHCGR gene, a diagnosis that was conducted rather tardily in the fourth decade of life. Our case expands the genotypic spectrum of LHCGR mutations and underlines the importance of thorough clinical and genetic examination, not only in pre- and post-pubertal children but also in adults originating from conservative socio-cultural backgrounds.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Richter-Unruh A, Verhoef-Post M, Malak S, et al, 2004 Leydig cell hypoplasia: absent luteinizing hormone receptor cell surface expression caused by a novel homozygous mutation in the extracellular domain. J Clin Endocrinol Metab 89: 5161-5167.
2. Richter-Unruh A, Martens JW, Verhoef-Post M, et al, 2002 Leydig cell hypoplasia: cases with new mutations, new polymorphisms and cases without mutations in the luteinizing hormone receptor gene. Clin Endocrinol (Oxf) 56: 103-112.
3. Martens JW, Verhoef-Post M, Abelin N, et al, 1998 A homozygous mutation in the luteinizing hormone receptor causes partial Leydig cell hypoplasia: correlation between receptor activity and phenotype. Mol Endocrinol 12: 775-784.
4. Themmen AP, Brunner HG, 1996 Luteinizing hormone receptor mutations and sex differentiation. Eur J Endocrinol 134: 533-540.
5. Ascoli M, Fanelli F, Segaloff DL, 2002 The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev 23: 141-174.
6. Richter-Unruh A, Korsch E, Hiort O, et al, 2005 Novel insertion frameshift mutation of the LH receptor gene: problematic clinical distinction of Leydig cell hypoplasia from enzyme defects primarily affecting testosterone biosynthesis. Eur J Endocrinol 152: 255-259.
7. Kossack N, Simoni M, Richter-Unruh A, et al, 2008 Mutations in a novel, cryptic exon of the luteinizing hormone/chorionic gonadotropin receptor gene cause male pseudohermaphroditism. PLoS Med 5: e88.
8. Segaloff DL, 2009 Diseases associated with mutations of the human lutropin receptor. Prog Mol Biol Transl Sci 89: 97-114.
9. Simoni M, 1998 Mutations of the G protein-coupled receptors of the hypothalamo-pituitary-gonadal axis. Where do we stand? Eur J Endocrinol 139: 145-147.
10. Mitre N, Lteif A, 2009 Treatment of familial male-limited precocious puberty (testot oxicosis) with anastrozole and bicalutamide in a boy with a novel mutation in the luteinizing hormone receptor. J Pediatr Endocrinol Metab 22: 1163-1167.
11. Xie YB, Wang H, Segaloff DL, 1990 Extracellular domain of lutropin/choriogonadotropin receptor expressed in transfected cells binds choriogonadotropin with high affinity. J Biol Chem 265: 21411-21414.
12. Fan QR, Hendrickson WA, 2005 Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433: 269-277.
13. Jiang X, Liu H, Chen X, et al, 2012 Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. PNAS 109: 12491-12496.
14. Braun T, Schofield PR, Sprengel R, 1991 Amino-terminal leucine-rich repeats in gonadotropin receptors determine hormone selectivity. EMBO J 10: 1885-1890.
16. Themmen APN, Huhtaniemi IT, 2000 Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev 21: 551-583.
15. Bruysters M, Verhoef-Post M, Themmen AP, 2008 Asp330 and Tyr331 in the C-terminal cysteine-rich region of the luteinizing hormone receptor are key residues in hormone-induced receptor activation. J Biol Chem 283: 25821-25828.
17. Nagasaki K, Katsumata N, Ogawa Y, et al, 2010 Novel C617Y mutation in the 7th transmembrane segment of luteinizing hormone/choriogonadotropin receptor in a Japanese boy with peripheral precocious puberty. Endocr J 57: 1055-1060.
Address for correspondence:
A.P. Athanasoulia; Endocrinology and Clinical Chemistry, Department of Internal Medicine, Max Planck Institute of Psychiatry, Kraepelinstrasse 2-10, 80804 Munich, Germany; Tel.: +498930622364, Fax: +4989306227460, E-mail: athanasoulia@mpipsykl.mpg.de
Received 23-04-2013, Accepted 16-09-2013