1Poznan University of Medical Sciences, 2nd Chair of Pediatrics, Department of Pediatric Endocrinology and Rheumatology, Molecular Endocrinology Laboratory, 2Karol Jonscher’s Clinical Hospital of Poznan University of Medical Sciences, Department of Pediatric Endocrinology and Rheumatology, Poznan, Poland
OBJECTIVE: X-linked Adrenal Hypoplasia Congenita (AHC) is a rare disorder caused by mutations in NR0B1 (DAX1) gene. DESIGN: We present two boys (cousins) with AHC who came to our attention at the age of 10 days and 15 days, respectively, in a life-threatening state. Laboratory studies in their neonatal periods showed hyponatremia and hyperkalemia. Primary adrenal insufficiency was confirmed, with severely low serum cortisol levels and high plasma ACTH levels. Hydrocortisone therapy with additional saline and glucose infusions were started immediately. Two exons of the NR0B1 (DAX1) gene were amplified using PCR and directly sequenced. RESULTS: Molecular analysis of the NR0B1 (DAX1) gene revealed a novel mutation, c.315G>A (p.W105X) in exon 1, resulting in the formation of a premature stop codon. Further studies showed that mothers, the maternal grandmother and two of six maternal great aunts were heterozygotes for the mutation. However, the mutation was absent in the maternal great-grandmother. CONCLUSIONS: We show that NR0B1 (DAX1) gene analysis is of great importance for the confirmation of the clinical diagnosis of AHC and highlights the role of genetic counseling for families of AHC patients. The absence of a somatic mutation in the great-grandmother suggests gonadal mosaicism as the mechanism for transmission of the NR0B1 (DAX1) mutation in this family.
Adrenal Hypoplasia Congenita (AHC), Adrenal insufficiency, NR0B1 (DAX1) gene
INTRODUCTION
Adrenal Hypoplasia Congenita (AHC, OMIM #300200) is a rare disorder that can be inherited in an X-linked or autosomal recessive pattern. In X-linked AHC, primary adrenocortical failure occurs due to the lack of the permanent adult cortical zone of the adrenals and is caused by loss-of-function mutations in the NR0B1 (DAX1) gene (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1) (OMIM*300473) located on the short arm of chromosome X (Xp21.3-p21.2) and encoding an orphan nuclear hormone receptor (transcription factor).1 It is expressed in the adrenals, gonads, hypothalamus and pituitary gland and controls the development and function of these tissues. NR0B1 (DAX1) protein functions as a dominant-negative regulator (transcriptional repressor) of the expression of other proteins involved in the adrenal steroidogenesis pathway, such as SF-1 (steroidogenic factor 1), steroidogenic acute regulatory protein (StAR), p450scc (cholesterol side chain cleavage enzyme) and 3β-hydroxysteroid dehydrogenase.2
NR0B1 (DAX1) plays a crucial role in the development of the adrenals and of the hypothalamic-pituitary-gonadal axis. NR0B1 (DAX1) gene disruption can result in various clinical phenotypes and may lead to congenital adrenal hypoplasia. This is characterized by salt loss early in life, hypoglycemia in early infancy or childhood,4 development of a life-threatening state, poor weight gain, emesis, prolonged postnatal jaundice, skin hyperpigmentation, hyponatremia, hyperkalemia, low cortisol level, low aldosterone level, high ACTH level and a higher plasma renin activity.5,6 The disorder is lethal unless appropriate steroid replacement therapy is quickly provided. Adrenal developmental failure is also characterized by low serum levels of adrenal androgens.7,8 Hypogonadotropic hypogonadism (HH) is the most frequently observed puberty disorder (absence or delayed puberty) caused by mutations in the NR0B1 (DAX1) gene and is due to impaired gonadotropin synthesis and release.1,9-11 The mechanism of the effect of NR0B1 (DAX1) gene action on puberty timing is still poorly understood, but it is known that NR0B1 (DAX1) acts at the levels of both the hypothalamus and the pituitary.12 This indicates the central role of NR0B1 (DAX1) transcription factor in the control of puberty timing.7,13-16 Normal puberty, peripheral precocious puberty, ACTH-dependent precocious puberty and central precocious puberty have also been reported so far, but many of these conditions are very rare.7,13-15 The mechanism by which NR0B1 (DAX1) induces normal adrenal and pubertal development still remains unknown.
Aim of the study
The aim of this study was to investigate the clinical and molecular background of congenital adrenal hypoplasia in two boys and their families, in whom a NR0B1 (DAX1) mutation was suspected.
CASE PRESENTATION
Patient 1. The phenotypic boy was born to non-consanguineous parents at 41 weeks of gestation with a birth weight of 3600 g. He was referred and admitted to our hospital on the 15th day of life from a Regional Hospital. Due to a suspicion of adrenal insufficiency he was treated with hydrocortisone. Physical examination revealed skin hyperpigmentation and a pericranial hematoma of the right parietal bone. He had well developed male external genitalia with an intrascrotal location of testes. Blood results showed hyponatremia (sodium 124 mEq/L) and hyperkalemia (6.06 mEq/L) with elevated ACTH at 688 pg/ml (normal <50) and a very low serum cortisol concentration (18 ng/ml), 17-hydroxyprogesterone (0.1 ng/ml), dehydroepiandrosterone sulfate (0.11 μmol/l) with no response to ACTH stimulation test, thus confirming primary adrenal failure. Abdominal ultrasound imaging revealed small adrenal glands (8-9 × 13-15 mm) – the right adrenal gland was described as hyperechogenic with a lacking cortex. A combination treatment with iv/oral hydrocortisone, oral fludrocortisone and supplementation with saline was continued.
Patient 2. The boy from a first uncomplicated pregnancy was born vaginally at 41 weeks (birth weight of 3270 g, Apgar 10). Both parents were healthy. The child was admitted to the hospital in a life-threatening state at the age of 10 days with a weight of 2990 g with a suspicion of adrenal failure. He was listless, weak, dehydrated and had a hyperpigmented and yellowish skin tone and very thin subcutaneous tissue. The hyperpigmentation of the skin, scrotum and nipples have been observed since birth. The boy also suffered from heart failure, pneumonia, jaundice and secondary anemia followed by an unexpected cardiac arrest. Blood results showed hyponatremia (sodium 112 mEq/L) and hyperkalemia (7.8 mEq/L) with elevated ACTH at 949.1 pg/ml (normal <50) and a very low serum cortisol concentration (7.8 ng/ml), 17-hydroxyprogesterone (0.17 ng/ml) with no response to ACTH stimulation test, thus confirming primary adrenal failure. Abdominal ultrasound was also performed and showed hypoplastic adrenal glands bilaterally (6-8 x 4-5 mm). Hydrocortisone therapy (iv/oral), fluid infusions, oral fludrocortisone, iron, folic acid and salt were started immediately.
Once primary adrenal insufficiency was confirmed, further testing was performed to diagnose X-linked AHC. Low levels of serum adrenal androgens (17-OHP, DHEA-S and androstendione) in both patients excluded 21-hydroxylase deficiency, the classic salt-wasting form of congenital adrenal hyperplasia (CAH). As part of the differential diagnosis of ACTH deficiency, congenital adrenal lipoid hyperplasia, familial glucocorticoid and ACTH resistance were also considered and excluded.
MATERIALS AND METHODS
Written informed consent was obtained from all individuals of the patients’ families and the study was approved by the local Ethics Committee (Nr 621/11). Molecular studies of the NR0B1 (DAX1) gene were performed using direct sequencing. Genomic DNA was isolated from peripheral blood leukocytes using QIAamp® DNA Blood Mini Kit (QIAGEN). Two sets of primers from Genomed (Poland) were used to amplify both exons of the NR0B1 (DAX1) gene. Polymerase chain reactions (PCR) were performed using HotStarTaq® DNA Polymerase (QIAGEN). PCR products were purified using QIAquick® Gel Extraction Kit (QIAGEN) and directly sequenced with the same primer pairs used in PCR. Sequencing reactions were performed with the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems) on an ABI Prism 3130XL Genetic Analyzer (Applied Biosystems) and the sequences were analyzed using VectorNTI9.0 Software (Invitrogen).
RESULTS
Direct sequencing of the NR0B1 (DAX1) gene revealed a novel mutation, c.315G>A (p.W105X) in exon 1 (Figure 1), that resulted in premature stop codon formation and thus elimination of the N-terminal domain of the NR0B1 (DAX1) protein.
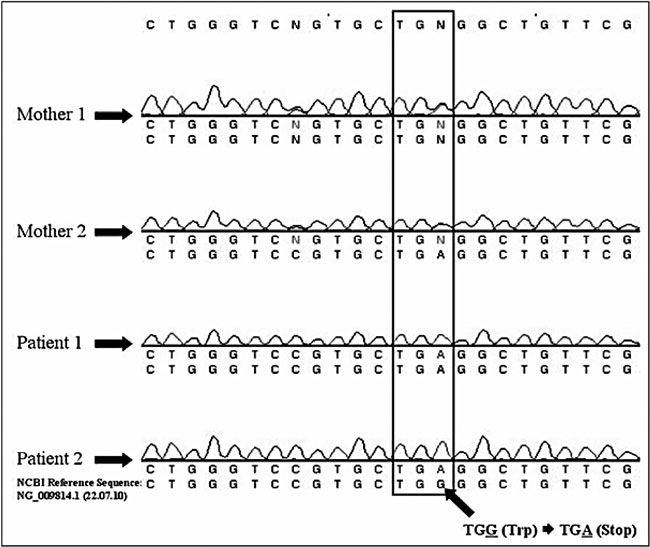
Figure 1. Identification of a novel c.315G>A; p.W105X nonsense mutation in patients 1 and 2 and their mothers. Chromatograms for other individuals tested in this study are not shown. The partial genomic sequence of the NR0B1 (DAX1) gene shows a p.W105X mutation at codon 105 by G to A substitution at nucleotide position 315. DNA sequences were compared to the NCBI Reference Sequence NG_009814.1 (22.07.2010).
This variant was not found in control samples of the current version of dataset ESP6500 deposited in the NHLBI Exome Variant Server, which is now comprised of a set of 2203 African-American and 4300 European-American, unrelated individuals, 6503 samples (13,006 chromosomes) in total (25.01.2013), and analysis using Mutation Taster (http://www.mutationtaster.org/) predicted the p.W105X mutation as a “disease causing” one.
Both mothers, whose menarche age was 14-15 and 14 years, respectively, the maternal grandmother and two of six maternal great aunts were heterozygotes for the mutation. However, the mutation was absent in the maternal great-grandmother (Figure 2).
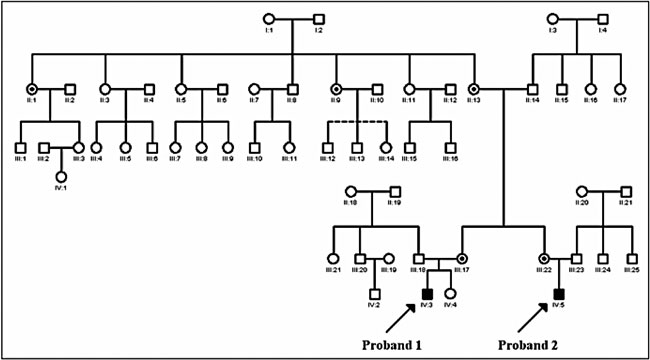
FIGURE 2. Pedigree of the family with a novel c.315G>A; p.W105X mutation in the NR0B1 (DAX1) gene. I:1, II:1, II:3, II:5, II:7, II:9, II:11, II:13, III:17, III:22, IV:3, IV:4 and IV:5 individuals were analyzed in this study. III:12, III:13 and III:14 individuals were adopted and thus were not included in molecular analysis. Two affected probands IV:3 (Patient 1) and IV:5 (Patient 2), healthy females, healthy males and female carriers (II:1, II:9, II:13, III:17 and III:22) of the mutation are indicated. The pedigree was prepared using GenoPro Software [◯ - healthy female, ⊙ - female carrier of the mutation, □ - healthy male, ▪- affected male].
DISCUSSION
In this study we present two boys with AHC caused by a novel mutation, c.315G>A (p.W105X) in exon 1 of the NR0B1 (DAX1) gene, whose clinical phenotype is unusual due to their normal testis and normal male external genitalia development. This mutation resulted in premature stop codon formation (TGA) in the codon encoding for tryptophane at position 105 and thus elimination of the N-terminal domain of NR0B1 (DAX1) protein.
Only one single nucleotide polymorphism G/C (SNP) at position 105 in NR0B1 (DAX1) is known (rs132630327: c.315G>C; p.W105C) that has clinical pathogenic significance.8 The c.315G>C; p.W105C mutation was identified in an 11-yr-old prepubertal Caucasian boy with mild AHC and isolated mineralocorticoid deficiency. The mutation was not detected in 100 controls, but was present in the patient’s mother as well as in his three asymptomatic male relatives, thus indicating phenotypic heterogeneity.8
The function of the aminoterminal part of NR0B1 (DAX1) protein still remains unclear, but it is known that this region is involved in protein-protein interactions with other molecules, such as SF-1 factor or estrogen receptor (ER), through its three, evolutionarily conserved LXXLL motifs.17,18 p.W105X mutation is located between the second and the third LXXLL motif. The tryptophane at position 105 is a highly conserved aminoacid in NR0B1 (DAX1) proteins and it is suggested that it is directly involved in NR0B1 (DAX1) structure and function. In vitro studies showed that there may be a correlation between the mutant NR0B1 (DAX1) activity and the severity of clinical phenotype when it comes to the onset of adrenal insufficiency, the severity of adrenal dysfunction and abnormalities with the biology of the reproductive system.16,19-21
c.314G>A (p.W105X) mutation, which is similar but not identical to the mutation identified in this study, was found only in one patient, described by Choi et al. in 2005, in whom adrenal hypoplasia congenita was diagnosed.3
Based on these two studies and our results, it may be suggested that W105 may be directly involved in NR0B1 (DAX1) structure and functions and codon 105 may be a hot spot for mutations. Our cases further expand the number of NR0B1 (DAX1) mutations reported up to now in the literature, as well as our clinical and molecular knowledge of AHC. Further functional research is, however, needed for clarification of the real impact of NR0B1 (DAX1) mutations on adrenal development and for explanation of the interaction between adrenals and the gonadotropin axis.
In our study, we also tested other members of the boys’ families and found that five of them were carriers of the mutation. However, we did not find the mutation in the boys’ great-grandmother (Figure 2), which may suggest gonadal mosaicism in her. One similar case of gonadal mosaicism and AHC has been described in the literature by Zhang et al. in a family with the 23-bp frameshift mutation 518del23bp.22
There is no clear evidence for a genotype-phenotype correlation between a mutation in NR0B1 (DAX1) and its structural consequences and the clinical phenotype. The age of onset of adrenal failure can vary within the same (but not in this) family, suggesting that other epigenetic factors influence the clinical course of AHC. This phenotypic heterogeneity may presumably be due to the effects of other genes modifying or compensating for NR0B1 (DAX1) function. Alternatively, environmental events such as illness or exposure to medications could unmask an underlying adrenal dysfunction. The variable expression of the AHC phenotype is reminiscent of other genetic disorders, particularly when the mutation has partial effects on protein function.
It is possible that specific mutations of the NR0B1 (DAX1) gene may also show atypical phenotypes, i.e., normal testis development, normal puberty and milder clinical phenotype of AHC, as in the boys with p.W105X mutation presented in this study, and which have been described by Choi et al.3
Primary adrenal insufficiency can occur at any age, in the neonatal period, in infancy or in childhood. Children usually present with failure to thrive or poor feeding, hypoglycemia, emesis, diarrhea, dehydration, shock and generalized skin pigmentation. Families harboring NR0B1 (DAX1) mutations often have a history of unexplained death of maternal male relatives. Most patients with X-linked AHC present with adrenal insufficiency early in life and have a combined glucocorticoid and mineralocorticoid deficiency.20 AHC is also characterized by puberty failure due to hypogonadotropic hypogonadism.12 Individuals with milder forms of AHC have also been described, but the incidence is not common. In such patients adrenal insufficiency is incomplete and their sexual maturation is delayed due to a mild gonadotropin deficiency or is arrested.16,19,21 These milder clinical phenotypes have incomplete loss-of-function of NR0B1 (DAX1) mutations.16,19,21 Boys with AHC may have episodes of elevated ACTH levels and therefore the question arises whether they are at risk of the same gonadal problem as CAH male patients, i.e., the risk of development of testicular adrenal rest tumors (TARTs).23,24 Development of the primitive adrenal cortex occurs close to the gonads, and TART is considered to be an aberrant adrenal tissue that has descended with the testes.25
On the other hand, boys with AHC and coexisting hypogonadotropic hypogonadism present a normal appearance of the testicles on ultrasound and also normal rete testis but they have azoospermia. The mechanism of azoospermia is gonadotropin-independent, since it was found that gonadotropin therapy was unsuccessful in induction of spermatogenesis. Perhaps NR0B1 (DAX1) directly regulates spermatogenesis in males and therefore they are resistant to gonadotropin treatment.26 Further observations of patients with the NR0B1 (DAX1) mutation, from early childhood through adolescence and into adulthood, can give us new insights into fertility aspects of these individuals.
CONCLUSIONS
The clinical signs and symptoms of AHC are similar to those observed in patients with the salt-wasting form of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency. It is very important to distinguish between these two disorders because they have a very different clinical course and different implications for genetic counseling. Molecular analysis of the NR0B1 (DAX1) gene, in uncertain cases, should be included as a routine diagnostic approach to differentiate both these disorders.27
The unique clinical state of the most likely carrier, the great-grandmother, suggests that she probably had gonadal mosaicism and that she most likely transferred the mutation to her three of six daughters (including the boys’ grandmother). The latter may suggest a 50% risk of transmission; however, the true mode of inheritance is unknown. We can only hypothesize that the great-grandmother had two types of germ cells, normal as well as mutated.
In summary, we identified a novel NR0B1 (DAX1) mutation in a family with two patients with severe AHC, while we also suggest that nonsense mutations in the aminoterminal region of NR0B1 (DAX1) may impair protein function and may thus cause the clinical presentation of AHC. The gonadal mosaicism in the great-grandmother may lie behind the further carriers and affected members in this family. Long-term follow-up of affected males with AHC is necessary, not only to ensure appropriate hormone replacement therapy but also to monitor other coexisting endocrine disturbances which may be observed, such as delayed/precocious puberty and infertility.
ACKNOWLEDGEMENTS
The authors are grateful to the patients and their families for their cooperation in this study, which was supported by funds of the Department of Pediatric Endocrinology and Rheumatology of Poznan University of Medical Sciences (Nr 502-01-01104118-06037 to Marek Niedziela) and by a Research Grant of the Department of Pediatric Endocrinology and Rheumatology of Poznan University of Medical Sciences (Nr 502-14-01104118-09627 to Aleksandra Rojek).
CONFLICT OF INTEREST
The authors have nothing to declare.
REFERENCES
1. Zanaria E, Muscatelli F, Bardoni B, et al, 1994 An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature 372: 635-641.
2. Iyer AK, McCabe ERB, 2004 Molecular mechanisms of DAX1 action. Mol Genet Metab 83: 60-73.
3. Choi J-H, Shin Y-L, Kim G-H, et al, 2005 Identification of novel mutations of the DAX-1 gene in patients with X-linked adrenal hypoplasia congenita. Hormone Res 63: 200-205.
4. Reutens AT, Achermann JC, Ito M, et al, 1998 Clinical and functional effects of mutations in the DAX-1 gene in patients with adrenal hypoplasia congenita. J Clin Endocrinol Metab 84: 504-511.
5. Achermann JC, Silverman BL, Habiby RL, Jameson JL, 2000 Presymptomatic diagnosis of X-linked adrenal hypoplasia congenita by analysis of DAX1. J Pediat 137: 878-881.
6. Nakae J, Abe S, Tajima T, et al, 1997 Three novel mutations and a de novo deletion mutation of the DAX-1 gene in patients with X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab 82: 3835-3841.
7. Wiltshire E, Couper J, Rodda C, Jameson JL, Achermann JC, 2001 Variable presentation of X-linked adrenal hypoplasia congenita. J Pediatr Endocrinol Metab 14: 1093-1096.
8. Verrijn Stuart AA, Ozisik G, de Vroede MA, et al, 2007 An amino-terminal DAX1 (NR0B1) missense mutation associated with isolated mineralocorticoid deficiency. J Clin Endocrinol Metab 92: 755-761.
9. Krude H, Biebermann H, Luck W, Horn R, Brabany G, Gruters A, 1998 Severe early- onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nature Genet 19: 155-157.
10. Muscatelli F, Strom TM, Walker AP, et al, 1994 Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 372: 672-676.
11. Vaidya B, Pearce S, Kendall-Taylor P, 2000 Recent advances in the molecular genetics of congenital and acquired primary adrenocortical failure. Clin Endocrinol 53: 403-418.
12. Habiby RL, Boepple P, Nachtigall L, Sluss PM, Crowley WF Jr, Jameson JL, 1996 Adrenal hypoplasia congenita with hypogonadotropic hypogonadism: Evidence that DAX-1 mutations lead to combined hypothalamic and pituitary defects in gonadotropin production. J Clin Invest 98: 1055-1062.
13. Peter M, Viemann M, Partsch CJ, Sippell WG, 1998 Congenital adrenal hypoplasia: Clinical spectrum, experience with hormonal diagnosis and report on new point mutations of the DAX-1 gene. J Clin Endocrinol Metab 83: 2666-2674.
14. Domenice S, Latronico AC, Brito VN, Arnhold IJP, Kok F, Mendonca BB, 2001 Adrenocorticotropin-dependent precocious puberty of testicular origin in a boy with X-linked adrenal hypoplasia congenita due to a novel mutation in the DAX1 gene. J Clin Endocrinol Metab 86: 4068-4071.
15. Loke K, Poh LK, Lee WW, Lai P, 2009 A case of X-linked adrenal hypoplasia congenita, central precocious puberty and absence of the DAX-1 gene: Implications for pubertal regulation. Horm Res 71: 298-304.
16. Mantovani G, Ozisik G, Achermann JC, et al, 2002 Hypogonadotropic hypogonadism as a presenting feature of late-onset X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab 87: 44-48.
17. Zhang Z, Thomsen JS, Johansson L, Gustafsson JA, Treuter E, 2000 DAX-1 functions as an LXXLL-containing corepressor for activated estrogen receptors. J Biol Chem 275: 39855-33989.
18. Suzuki T, Kasahara M, Yoshioka H, Morohashi K, Umesono K, 2003 LXXLL-related motifs in DAX-1 have target specificity for the orphan nuclear receptors AD4BP/SF-1 and LRH-1. Mol Cell Biol 23: 238-249.
19. Tabarin A, Achermann JC, Recan D, et al, 2000 A novel mutation in DAX1 causes delayed-onset adrenal insufficiency and incomplete hypogonadotropic hypogonadism. J Clin Invest 105: 321-328.
20. Achermann JC, Meeks JJ, Jameson JL, 2001 Phenotypic spectrum of mutations in DAX-1 and SF-1. Mol Cell Endocrinol 185: 17-25.
21. Ozisik G, Mantovani G, Achermann JC, et al, 2003 An alternate translation initiation site circumvents an amino-terminal DAX1 nonsense mutation leading to a mild form of X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab 88: 417-423.
22. Zhang Y-H, Guo W, Wagner RL, et al, 1998 DAX1 mutations map to putative structural domains in a deduced three-dimensional model. Am J Hum Genet 62: 855-864.
23. Claahsen-van der Grinten HL, Otten BJ, Hermus AR, et al, 2008 Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia can cause severe testicular damage. Fertil Steril 89: 597-601.
24. Niedziela M, Talarczyk J, Jedrzejczak P, 2012 Testicular adrenal rest tumors (TARTs) as a male infertility factor. Case report. Ginekol Pol 83: 700-702.
25. Barwick TD, Malhotra A, Webb JA, Savage MO, Reznek RH, 2005 Embryology of the adrenal glands and its relevance to diagnostic imaging. Clin Radiol 60: 953-959.
26. Mantovani G, De Menis E, Borretta G, et al, 2006 DAX1 and X-linked adrenal hypoplasia congenita: Clinical and molecular analysis in five patients. Eur J Endocrinol 154: 685-689.
27. Rojek A, Obara-Moszynska M, Malecka E, Slomko-Jozwiak M, Niedziela M, 2013 NR0B1 (DAX1) mutations in patients affected by congenital adrenal hypoplasia with growth hormone deficiency as a new finding. J App Genet 54: 225-230.
Address for correspondence:
Aleksandra Rojek, University of Medical Sciences in Poznan, 2nd Chair of Pediatrics, Department of Pediatric Endocrinology and Rheumatology, Molecular Endocrinology Laboratory, 27/33 Szpitalna Street, 60-572 Poznan, Poland, Tel.: +48 61 8491 424, Phone/Fax: +48 61 8480 291,
E-mail: aleksandra.rojek@gmail.com
Received 27-03-2013, Accepted 28-06-2013