1Adolescent Medicine, I Pediatric Division, Department of Obstetrics, Gynaecology and Pediatrics; 2Division of Pediatric Oncology, Department of Obstetrics, Gynecology and Pediatrics; 3Laboratory of Medical Genetics, Department of Laboratory Medicine; University Hospital, Pisa, Italy
BACKGROUND: Short stature represents one of the main features of children with Noonan syndrome. The reason for impaired growth remains largely unknown. OBJECTIVE: To assess GH and IGF1 secretion in children with Noonan syndrome. Patients: 12 prepubertal children with Noonan syndrome due to mutations in the PTPN11 gene [7 males, 6 females; median age, years: 8.6 (range 5.1-13.4)] were studied; 12 prepubertal children with short stature (SS) [7 males, 5 females; median age, years: 8.1 (range 4.8-13.1)] served as the control group. Measurements: GH secretion after arginine stimulation test; IGF1 generation test by measurement of IGF1 levels before and after recombinant GH (rGH) administration (0.05 mg/kg/day for 4 days). RESULTS: Baseline and stimulated peak values of GH were not significantly different between the two groups. At +120 minutes, GH levels remained significantly higher (p = 0.0121) in comparison with baseline values in children with Noonan syndrome. Baseline IGFI levels in patients and in SS controls were not significantly different, in contrast to values after the rGH generation test [205 ng/mL (interquartiles 138.2-252.5 ng/mL) and 284.5 ng/mL (interquartiles 172-476 ng/mL), respectively; p = 0.0248]. IGF1 values were significantly related to height (baseline: r = 773, p = 0.0320; peak: r = 0.591, p = 0.0428) in children with Noonan syndrome. CONCLUSIONS: Blunted increase of IGF1 after the rGH generation test was present in children with Noonan syndrome due to mutations in the PTPN11 gene in comparison with SS children. This finding may be due to partial GH resistance in the former likely related to altered Ras-MAPK signaling pathway.
Growth hormone, IGF1 generation test, Noonan syndrome, PTPN11 gene, Short stature
Introduction
Noonan syndrome (OMIM 163950, incidence 1:1000 - 1:2500 live births) is a heterogeneous genetic disorder characterized by typical dysmorphic facial features, chest deformities, congenital heart disease, mild mental retardation, cryptorchidism in males and clotting disorders.1,2 In 30-60% of the patients, the syndrome is related to mutations in the PTPN11 gene, which encodes the non-receptor protein tyrosine phosphatase SHP-2; this protein is involved in a variety of intracellular signal cascades downstream of receptors for growth factors, cytokines and hormones.3,4 The mutations associated with Noonan syndrome result in a gain of function of SHP-2 and determine derangements of the post-receptorial Ras-MAPK signaling pathway.3-5 Other genes involved in the Ras-MAPK signaling cascade have been identified in patients without PTPN11 mutations.1,2
Short stature is a cardinal feature of the syndrome.1,6 Adult height is about 161-170 cm for males and 150-153 cm for females and it is below third percentile in about 40% of males and 55% of females.1,7-9,
The cause of growth impairment in Noonan syndrome remains poorly understood. Some studies demonstrated subnormal stimulated GH levels in a minority of the investigated patients,1,10,11 while other authors reported normal GH response or tendency to increased GH peaks.12-15 The spontaneous GH secretion demonstrated qualitatively abnormal secretion with high trough GH concentrations.13,16 The levels of insulin-like growth factor-I (IGF-I) have been found to be reduced,13-16 suggesting that partial GH resistance may be operative in children with Noonan syndrome.12,14
The IGF-I generation test is a dynamic test to assess the sensitivity to GH through the measurement of serum IGF-I prior to and after the administration of recombinant GH (rGH).17-19
In this paper, we report on GH secretion assessment and the IGF-I generation test in prepubertal children with Noonan syndrome due to mutations in the PTPN11 gene. Results are compared with those obtained in a group of prepubertal subjects with short stature.
Patients and methods
Patients
Twelve prepubertal children [7 males (2 brothers) and 5 females; median age, years: 8.6 (range 5.1 - 13.4)] with Noonan syndrome were studied. All the children were born at term (39.9 ± 0.7 weeks) after uneventful pregnancies (birth weight, g: 3198 ± 363; birth length, cm: 49.7 ± 1.4). The individual clinical data are summarized in Table 1. All the subjects presented short stature and every child showed a height SDS below mid-parental height SDS. Heterozygous PTPN11 mutations were found in all children (Table 1). Cardiac anomalies are present in 6/12 (Table 1); median height in patients with cardiac anomalies was not significantly different (p = 0.749) from that of children with normal cardiac morphology [-2.95 SDS (interquartiles -3.26, -1.95 SDS) and -2.75 SDS (interquartiles -3.08, -1.96 SDS), respectively]. No patients were prescribed cardiologic drugs when GH and IGFI status were assessed. Two boys were brothers, both without cardiac involvement (Table 1). Twelve prepubertal children [median age, years: 8.1 (range 4.8 - 13.1); 7 males, 5 females; two brothers] with short stature (SS) [height, SDS: median -2.4 (interquartiles –3.8, -1.4); bone age, years: median 7.3 years (interquartiles 4.2, 10)] were recruited as the control group. Extensive clinical evaluation by expert pediatric endocrinologists (SB, GIB) and a clinical genetist (BT) did not reveal signs of Noonan syndrome or other disorders associated with impaired growth. History of familial short stature was present in 5/12.
In both groups, the occurrence of occult celiac disease or other chronic gastrointestinal disorders was ruled out by appropriate biochemical analysis; reactive C protein, thyroid values, renal function and albumin levels were normal. Body mass index (BMI) was in the normal range and not significantly different between children with Noonan syndrome and SS controls [median BMI, kg/m2: 15.7 (interquartiles 14.9, 16.4) and 15.3 (interquartiles 14.1, 18.1), respectively)]. None of the subjects showed clinical and/or biochemical signs of pubertal development [pubertal stage Ph1 and B1 or G1, for females and males, respectively; prepubertal levels of LH and 17β-estradiol (<75 pmol/L) or testosterone (<1 nmol/L)]. Informed parental consent and approval by our department ethics committee were obtained before the study.
Auxological assessment
Height was measured by standard methodology using a wall-mounted stadiometer and calculating the average of three measurements. Auxological data were transformed to standard deviation scores (SDS) using Italian reference values.20 Mid-parental height was calculated as follows: (measured father’s height + measured mother’s height)/2 + 6.5 cm for boys and – 6.5 cm for girls. Bone age was assessed by the Greulich and Pyle method.21
Hormonal assessment
GH secretion in response to the arginine provocative test (0.5 mg/kg/i.v. over a 30-minute period) was performed after an overnight fast in children with Noonan syndrome and controls.22 Serum samples for determination of GH were collected at 0, 30, 60, 90 and 120 minutes. A normal response was defined as a GH peak above 10 ng/ml.22,23
The IGFI generation test was performed at least two weeks after the GH stimulation test. A blood sample was collected in the morning after 12 h of fasting; then, rGH was administered in the evening (h. 8.00 p.m.) for four consecutive days (0.05 mg/kg/day).17,18 A second blood sample was taken in the morning of the fifth day.17,18 A positive response to the IGF-I generation test was defined as a Δ-increase of serum IGF-I levels >15 ng/mL over the baseline in the samples collected in the morning of the fifth day after 4 rGH doses.16,17 The biochemical data used were the absolute laboratory value obtained at each serum sample and the Δ between the basal sample and the one after rGH. Data in the patients were compared with those of controls. All serum samples were kept at -80°C up to IGF1 assessment, after which they were processed in the same assay batch.
Serum GH was determined by RIA (Spectria HGH, Orion Diagnostica, Espoo, Finland). The assay was standardized against the WHO 1st international reference preparation for hGH (WHO IRP 66/217)in which 1 mg = 1.6 IU of hGH; sensitivity was 0.6 ng/ml; levels from 2 to 23 ng/ml presented a CV intra-assay <8% and levels from 4 to 14 ng/ml presented a CV inter-assay <6%. Serum IGF-1 was measured with an IRMA kit (IGF-1-RIACT, Cis Bio, Gif-sur-Yvette, France) which uses two monoclonal antibodies directed against two different epitopes of the IGF-1 molecule. No extraction step was performed, but the bound IGF-1 was displaced from IGFBPs by acidification. A large excess of IGF-II was then added to the acid-treated serum to prevent re-association of IGF-1 with its carrier proteins when buffer was added. The limit of detection, defined as being the smallest detectable concentration different from zero with a probability of 95%, was 1 ng/ml. Intra-assay CV and inter-assay CV were <5% and <8%, respectively. Internal reference values for IGF1 (277 ± 93 ng/ml) were obtained in healthy prepubertal children (n = 35, age 8.9 ± 2.4 years, 16 females, 14 males; height within 25-75° centile for chronological age; body weight within +10% of ideal body weight for height); they were free from endocrine and non-endocrine diseases affecting serum IGF1 values.
Molecular analysis
Blood samples were taken in ethylenediamine tetraacetate (EDTA)-containing tubes and DNA was extracted by BioRobot EZ1 (Qiagen).
The PTPN11 gene coding regions and exon-intron boundaries were amplified by PCR. Purified PCR products were bidirectionally sequenced using the ABIPrism BigDye Terminator v3.1 Cycle Sequencing kit (Applera, Italy) and, after removal of unincorporated dye terminators, the sequencing reactions were separated on an ABIPrism 3130XL Genetic Analyzer (Applera, Italy) and analyzed by means of DNASequencingAnalysis and SeqScape software.
Statistical analysis
Results are expressed as median and interquartiles or as mean ± SD, when specified. Non-parametric statistical analysis was used to compare the data. The relationships between pairs of variables were assessed by the Wilcoxon rank-sum test and by repeat analysis of variance, when appropriate. A “p” value less than 0.05 was considered to be significant in all statistic analysis. Statistics were obtained by using GraphPad InStat, version 3.10.
Results
GH secretion
After arginine stimulation, a GH peak above the cut-off value of 10 ng/mL was found in all the children with Noonan syndrome and in SS controls. Baseline and stimulated peak values of GH were not significantly different between the two groups (Figure 1). At the end of the provocative test (+120 minutes), GH levels were not significantly different in comparison with baseline values in SS children, while they remained significantly higher in patients with Noonan syndrome, this demonstrating a significant difference between the two groups (Figure 1).

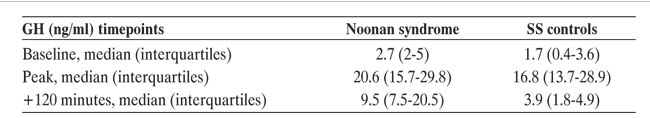
Figure 1. GH levels (mean ± SD) in children with Noonan syndrome (•) and in SS children (○) during the arginine stimulation test
IGF1
Baseline IGF1 levels in patients and in SS controls were significantly reduced (p = 0.0000 and p = 0.0002, respectively) in comparison with internal reference values, while significant differences between the two groups were not found (Figure 2). After the rGH generation test, 11/12 of the patients and all SS children showed incremental values above the cut-off limit of 15 ng/ml. However, the absolute and SDS peak levels of IGF1 in patients were significantly reduced in comparison with those of controls (Figure 2). Δ-increment in patients was significantly lower than that in controls (Figure 2). After rGH administration, serum IGF1 SDS levels remained significantly low in children with Noonan syndrome in comparison with reference values (p = 0.0046), while they normalized in controls (p = 0.2070). Height was significantly related to baseline and stimulated IGF1 levels in children with Noonan syndrome (basal absolute values: r = 0.773, p = 0.032; basal SDS; r = 0.772, p = 0.032; stimulated absolute values: r = 0.591, p = 0.0428; stimulated SDS: r = 0.595, p = 0.0411). No other significant relationships were found (data not shown).
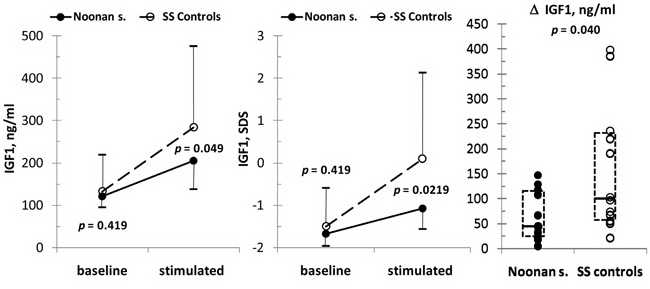
Figure 2. Baseline and stimulated levels (median and interquartiles) of IGF1 (absolute values: panel A; SDS values: panel B) in children with Noonan syndrome (•) and in SS children (○). In panel C, individual absolute D-increment of IFG1 after rGH administration in the two groups is reported; the dotted box represents median and lower/upper interquartiles.
Discussion
Short stature is a well known feature of children and adults with Noonan syndrome.6-9 Although height and weight are usually in the normal range at birth, as in this patient sample, height drops off during the first months of extrauterine life, leading to a decline in growth curve.1 The biological basis of impaired postnatal growth in Noonan syndrome is not completely understood.1 Assessment of GH secretion demonstrated inconclusive results. Some papers suggested GH insufficiency or GH neurosecretory dysfunction.10-12 Other authors have reported abnormalities in GH secretion unrelated to the auxological findings.16 However, since several studies involved patients who were not characterized by molecular analysis, they possibly included subjects of different genetic backgrounds.
In the present study, well genetically defined prepubertal patients were investigated and data were compared with those obtained in the SS controls. We found a GH peak above 10 ng/mL in all children. Although a single GH stimulatory test was performed, this result, in agreement with other studies,13-15 was not consistent with classical GH deficiency22. Furthermore, adequate response to one stimulatory test indicates that the child’s GH secretory status is not impaired.23 A tendency to higher GH peak and significantly higher GH values at the last timepoint of the arginine stimulatory test was found in Noonan children, this corroborating that a mutated SHP2 protein may interfere with intracellular GH signaling.12 More sensitive protocols in larger numbers of patients should be carried out in order to reach definitive conclusions.
Despite normal GH response to the arginine provocative test, baseline IGF1 levels were significantly reduced in patients with Noonan syndrome. Decreased IGF1 values have previously been reported,13-15 although some authors did not confirm this finding.24 To further explore the GH-IGF1 axis, we performed the IGF1 generation test. Because only prepubertal patients were studied, confounding factors related to the pubertal IGF1 increase were avoided.25 In addition, BMI values were similar in the two groups, ruling out the possibility that the different IGF1 pattern may be related to BMI differences. An adequate response was found in the majority of our patients (90%), according to the cut-off value of 15 ng/ml.17-19 The unresponsive girl did not show peculiar clinical features, but she had the highest GH peak after arginine stimulation (patient 1). Jorge et al also reported a boy with Noonan syndrome, diagnosed on the basis of clinical findings, who failed to respond to rGH stimulation, but he presented normal response to a second test.26 Although this patient’s data may indicate lack of reproducibility of the IGFI generation test and point to the need to perform a second test to confirm the GH insensitivity,26 we were unable to do this in our unresponsive girl.
The results of the IGF1 generation test demonstrated that the majority of our children with Noonan syndrome did not present classic biochemical GH insensitivity.19,27 However, the absolute and Δ-increment IGF1 values in this patient group was significantly reduced in comparison with that observed in the SS children. In the healthy children, the reported mean Δ-increment of IGF1 with the rGH dose we used was 322 ng/mL19 and 144.8 ng/mL.18 Though these results exhibited a great variability of the IGF1 generation test, they are consistent with the Δ-increment we found in the SS controls, while the mean increase in patients with Noonan syndrome was 63.9 ng/mL. This finding suggests an impaired responsiveness to GH stimulation and is in keeping with the lower IGF1 increase14 as well as the lower growth velocity reported in patients with Noonan syndrome positive for PTPN11 mutations during rGH replacement therapy by contrast to those not sharing the mutation.13,15
Recently, a girl with Noonan-like syndrome with loose anagen hair (OMIM 607721) has been described in whom severe short stature was associated with blunted IGF1 increase during a generation test performed by using a different rGH dose (0.33 µg/kg/die for 4 days).28 Ras-MAPK signal dysregulation is also operative in this syndrome.29 Taken together, these data seem to imply that all genetic alterations involving the Ras-MAPK signaling cascade may determine partial GH insensitivity.28
Major limitations of our study were the small group of patients and the overlapping IGF1 values between patients and SS controls due to the large variability in IGF1 increase. Other GH doses has been proposed to investigate partial defects in GH response.17-19 Thus, lower GH doses and a different sampling schedule could be useful in future studies to better delineate the pattern of the GH-IGF1 axis derangement in children with Noonan syndrome and PTPN11 mutations. In addition, adequate cohorts of patients of different genetic backgrounds should be investigated to assess the effects of the various genes involved in cell signaling through the Ras-MAKP pathway on GH sensitivity. Certainly, there is a need for such studies to be carried out based on stricter selection of patients in order to pinpoint those more likely to benefit from treatment with GH or IGFI. Finally, the effect of different PTPN11 mutations on the GH resistance phenotype should be assessed by in vitro functional studies.
REFERENCES
1. Padidela R, Camacho-Hübner C, Attie KM, Savage MO, 2008 Abnormal growth in Noonan syndrome: genetic and endocrine features and optimal treatment. Horm Res 70: 129-136.
2. van der Burgt I, 2007 Noonan syndrome. Orphanet J Rare Dis 2: 4.
3. Tartaglia M, Gelb BD, 2005 Noonan syndrome and related disorders: genetics and pathogenesis. Ann Rev Genom Hum Genet 6, 45-68.
4. Sarkozy A, Digilio MC, Marino B, Mingarelli R, Tartaglia M, Dalla Piccola B, 2006 Noonan’s syndrome and related disorders: clinical molecular update and guidelines. Ital J Pediatr 32: 45-155.
5. Neel BG, Gu H, Pao L, 2003 The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sciences 28: 284-293.
6. Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA, 2007 The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child 92: 128-132.
7. Ranke MB, Heidemann P, Knupfer C, Enders H, Schmaltz AA, Bierich JR, 1988. Noonan syndrome: growth and clinical manifestations in 144 cases. Eur J Pediatr 148: 220-227.
8. Noonan JA, Raaijmakers R, Hall BD, 2003 Adult height in Noonan syndrome. Am J Med Genet A 123A: 68-71.
9. Smpokou P, Tworog-Dube E, Kucherlapati RS, Roberts AE, 2012 Medical complications, clinical findings, and educational outcomes in adults with Noonan syndrome. Am J Med Genet A 158A: 3106-3111.
10. Cotterill AM, McKenna WJ, Brady AF, et al, 1996 The short-term effects of growth hormone therapy on height velocity and cardiac ventricular wall thickness in children with Noonan’s syndrome. J Clin Endocrinol Metab 81: 2291-2297.
11. Romano AA, Blethen SL, Dana K, Noto RA, 1996 Growth hormone treatment in Noonan syndrome: the National Cooperative Growth Study experience. Pediatrics 128: S18-21.
12. Tanaka K, Sato A, Naito T, Kuramochi K, Itabashi H, Takemura Y, 1992 Noonan syndrome presenting growth hormone neurosecretory dysfunction. Int Med 31: 908-911.
13. Binder G, Neuer K, Ranke MB, Wittekindt NE, 2005 PTPN11 mutations are associated with mild growth hormone resistance in individuals with Noonan syndrome. J Clin Endocrinol Metab 90: 5377-5381.
14. Limal JM, Parfait B, Cabrol S, et al, 2006 Noonan syndrome: relationships between genotype, growth, and growth factors. J Clin Endocrinol Metab 91: 300-306.
15. Ferreira LV, Souza SA, Arnhold IJ, Mendonca BB, Jorge AA 2005 PTPN11 (protein tyrosine phosphatase, nonreceptor type 11) mutations and response to growth hormone therapy in children with Noonan syndrome. J Clin Endocrinol Metab 90: 5156-5160.
16. Noordam C, van der Burgt I, Sweep CG, Delemarre-van de Waal HA, Sengers RC, Otten BJ, 2001 Growth hormone (GH) secretion in children with Noonan syndrome: frequently abnormal without consequences for growth or response to GH treatment. Clinical Endocrinol (Oxf) 54: 53-59.
17. Blum F, Cotterill AM, Postel-Vinay MC, Ranke MB, Savage MO, Wilton P, 1994 Improvement of diagnostic criteria in growth hormone insensitivity syndrome: solutions and pitfalls. Pharmacia study group on insulin-like growth factor I treatment in growth hormone insensitivity syndromes. Acta Paediatrica Suppl 399: 117-124.
18. Pessoa de Queiroz AN, Collett-Solberg PF, Cardoso ME, Jusan RC, Vaisman M, Guimarães MM, 2007 IGF-I, IGFBP-3 and ALS generation test in Turner syndrome. Growth Hormone & IGF Research 17: 254-260.
19. Selva KA, Buckway CK, Sexton G, et al, 2003 Reproducibility in patterns of IGF generation with special reference to idiopathic short stature. Horm Res 60: 237-246.
20. Cacciari E, Milani S, Balsamo A, et al, 2006 Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J Endocr Invest 29: 581-593.
21. Greulich WW, Pyle SI, 1959 Radiographic atlas of skeletal development of the hand and wrist. 2nd ed. Stanford, CA: Stanford University Press.
22. Carrillo AA, Bao Y, 2007 Hormonal dynamic tests and genetic tests used in pediatric endocrinology. In “Pediatric Endrocrinology”, 5th ed, ch 33, pp 737-767. ed Lifshitz F. New York, NY: Informa Healthcare USA, Inc.
23. Cappa M Loche S, 2005 Clinical and laboratory evaluation of short statured children. Ital J Pediatr 31: 26-32.
24. De Lima Jorge AA, 2007 Diagnosis and long-term human growth hormone treatment of a boy with Noonan syndrome. Horm Res 67: Suppl 1: 98-101.
25. Juul A, Bang P, Hertel NT, et al, 1994 Serum insulin-like growth factor-I in 1030 healthy children, adolescents, and adults: relation to age, sex, stage of puberty, testicular size, and body mass index. J Clin Endocrinol Metab 78: 744-752.
26. Jorge AA, Souza SC, Arnhold IJ, Mendonca BB, 2002 Poor reproducibility of IGF-I and IGF binding protein-3 generation test in children with short stature and normal coding region of the GH receptor gene. J Clin Endocrinol Metab 87: 469-472.
27. Rosenfeld RG, Belgorosky A, Camacho-Hubner C, Savage MO, Wit JM, Hwa V, 2007 Defects in growth hormone receptor signaling. Trends Endocrinol Metab 18: 134-141.
28. Capalbo D, Melis D, De Martino L, et al, 2012 Noonan-like syndrome with loose anagen hair associated with growth hormone insensitivity and atypical neurological manifestations. Am J Med Genet A 158A: 856-860.
29. Cordeddu V, Di Schiavi E, Pennacchio LA, et al, 2009 Mutation of SHOC2 promotes aberrant protein Nmyristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 41: 1022-1026.
Address for correspondence:
Silvano Bertelloni, MD, I Pediatric Division, Department
of Obstetrics, Gynecology and Pediatrics, Santa Chiara
University Hospital, Via Roma, 67, 56126 - Pisa, Italy,
Tel.: +39 050 992 743; Fax: +39 050 993 044; e-mail:
s.bertelloni@ao-pisa.toscana.it
Received 08-09-12, Accepted 25-01-12