1"C.I. Parhon" National Institute of Endocrinology, 2"Carol Davila" University of Medicine and Pharmacy, Bucharest, Romania
Primary hyperparathyroidism is a common endocrine disorder that is mostly caused by solitary tumors within the parathyroid glands. Characterized by early debut and higher frequency of multiple parathyroid masses, familial forms of primary hyperparathyroidism are caused by the already known mutations of: menin (MEN1 syndrome), RET proto-oncogene (MEN2 syndrome), HRPT2-parafibromin (hyperparathyroidism-jaw tumor syndrome), calcium sensing receptor gene (familial hypocalciuric hypercalcemia). A specific mutation in FIHP has not been identified in the majority of affected families. Recent studies revealed menin, HRPT2 and calcium-sensing receptor mutations in patients with FIHP. Whether FIHP is a variant or an early stage of MEN1 syndrome or hyperparathyroidism-jaw tumor syndrome is yet to be established. We present three siblings with familial isolated hyperparathyroidism due to solitary parathyroid adenoma and favorable evolution post-parathyroidectomy. Genetic tests revealed HRPT2 mutation.
Familial isolated hyperparathyroidism, HPT-JT, HRPT2
INTRODUCTION
Familial isolated hyperparathyroidism (FIHP) is a rare hereditable disorder characterized by hypercalcemia, high parathyroid hormone (PTH) level and isolated parathyroid tumors. FIHP was suggested as being a syndrome clinically and genetically distinct from multiple endocrine neoplasia (MEN), familial hypocalciuric hypercalcemia and hyperparathyroidism-jaw tumor syndrome.
CASE REPORTS
We present below a family with isolated HPT. Affected members are the father and the two sons (Figure 1).

Figure 1. Pedigree.
Case 1
The younger son (23 years) was the first to be diagnosed. He complained of progressive bone pain and marked reduction of muscle force.
Laboratory data (Table 1) revealed hypercalcemia, hypophosphatemia and inappropriately high parathyroid hormone, suggesting primary hyperparathyroidism. There was no evidence of other endocrine neoplasia.
Cervical ultrasound showed a nodule in the lower posterior right thyroid lobe (Figure 2).
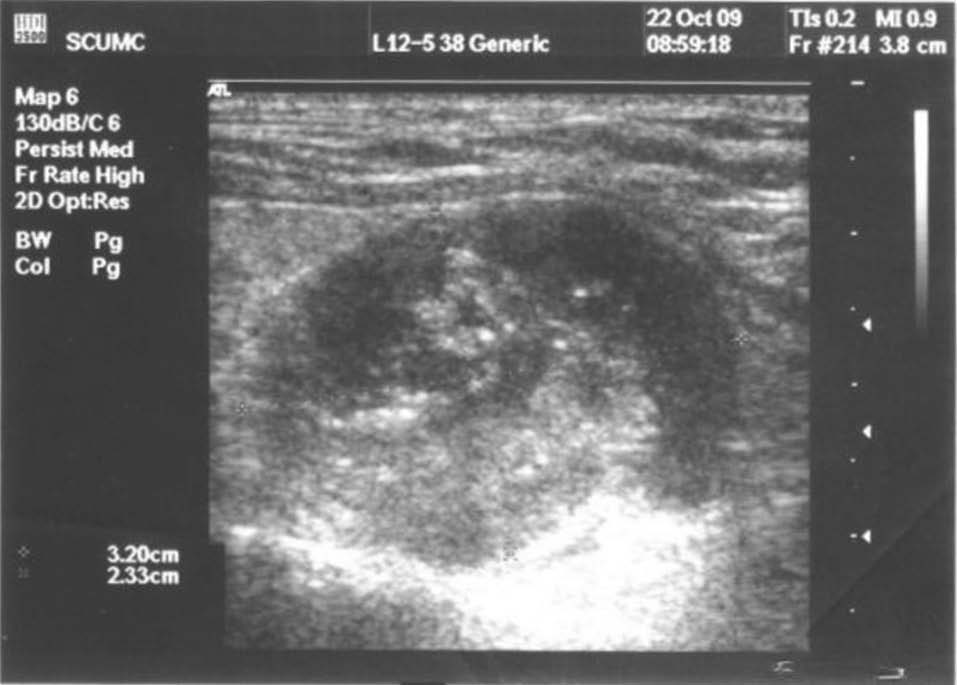
Figure 2. Cervical ultrasound: 26/23mm nodule in right thyroid lobe, located posteriorly, with mixed structure and intense vascularization.
Technetium scintigraphy showed increased uptake in the lower pole of the right thyroid lobe. CT scan showed a 28/20mm heterogenous nodule on the right thyroid lobe, confirming it as a potential parathyroid adenoma. We also found osteitis fibrosa cystica i.e. multiple osteolytic bone lesions with intact periosteum located on the clavicle, sternum, bilateral humerus, scapulas, ribs and pelvic bones. The largest (67/34mm) lesion occupied almost entire cortex at the level of the left anterior sacral wing.
Preoperative management consisted of intravenous infusion of 1.5 liters of saline solution 0.9%, 90mg of Pamidronate and 100UI of Calcitonin 3 times a day for 3 days. Right inferior parathyroidectomy was performed and the histopathological report confirmed the presence of a parathyroid adenoma with oxyphil cells, old hemorrhagic areas, areas of cholesteatoma and impregnation with calcareous matter and capsular sclerosis. Unexpectedly, postoperative calcemia and PTH level remained elevated (10-11mg/dl, respectively, 100pg/ml) but spontaneously normalized after 3 months.
Case 2
The second patient was the 55-year old father, who had a history of high blood pressure (170/100mmHg) and calcium oxalate nephrolithiasis. His blood tests revealed primary hyperparathyroidism (Table 1). Thyroid ultrasound showed a 40/30mm transonic nodule with floating echoes inside the lower left thyroid lobe and another 8mm nodule in the lower posterior right lobe. Technetium scintigraphy showed increased uptake in the lower pole of the left thyroid lobe (Figure 3).
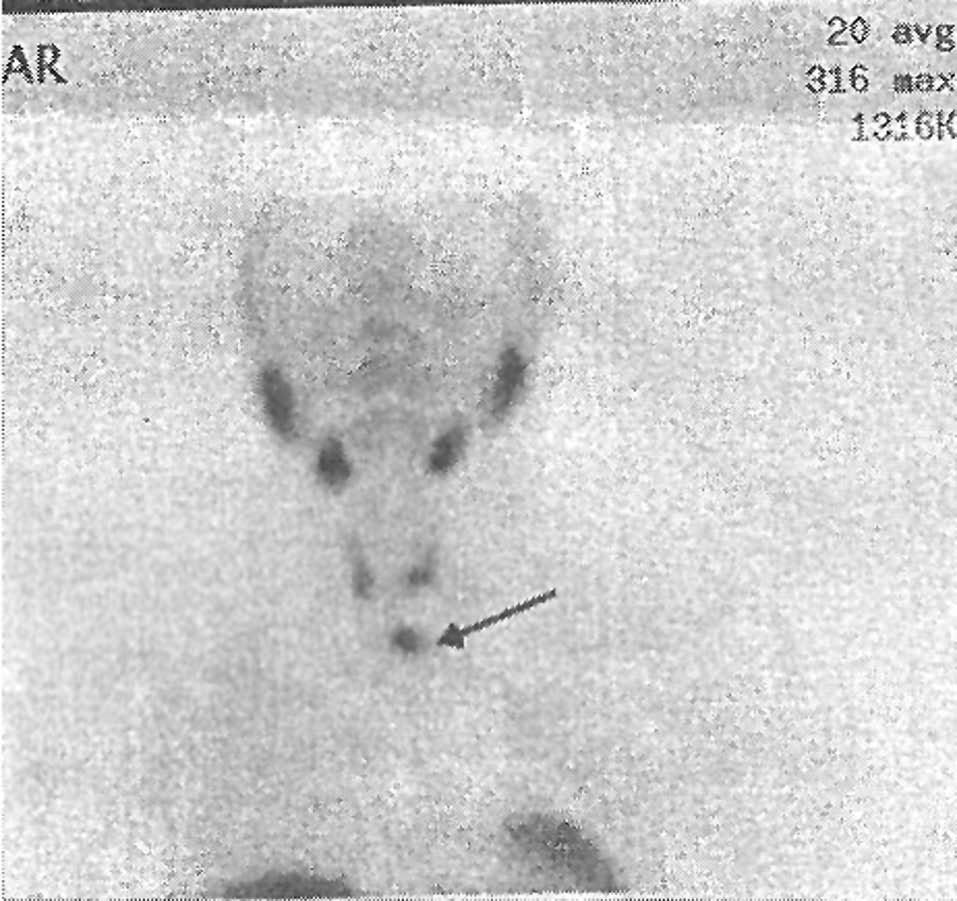
Figure 3. Technetium scintigraphy: increased uptake in lower pole of left thyroid lobe.
Cervico-mediastinal CT scan showed in the prevascular space, in intimate contact with the left thyroid lobe, a 35/33mm nodule with mixed structure well defined by a thin and iodophilic capsule.
Left inferior parathyroidectomy and right hemithyroidectomy were performed. The histopathological report confirmed the presence of a parathyroid adenoma with oxyphil cells, sclerous capsule and septa and areas of cholesteatoma. Postoperative evolution was similar to that of Case 1 with persistent hypercalcemia and elevated PTH, spontaneously normalized after 3 months. Thyroid replacement therapy with 50μg LT4/day was prescribed.
Case 3
The last member of the affected family is the eldest son (31 years). He has a history of repeated incidence of renal colic and diffuse bone pain. Laboratory data revealed primary hyperparathyroidism (Table 1).
Cervical ultrasound revealed: two nodules in the middle and lower part of the right thyroid lobe (Figure 4).

Figure 4. Thyroid ultrasound: in right thyroid lobe a 20/30mm mixed nodule and a 7/18mm hypodense nodule, both intensely vascularized.
Technetium scintigraphy showed increased uptake in the lower right thyroid lobe. CT scan indicated the same 2 nodules as suspected parathyroid lesions: superior = 26/23/30mm and inferior = 17/18/14mm.
Renal ultrasound revealed the presence of a 14mm stone in the right kidney.
DXA scan indicated loss of cortical bone:
– L1-L4: BMD = 1.037; T = -1.5; Z = -1.3
– femoral neck: BMD = 0.757; T = -2.4; Z = -2.3
– radius: BMD = 0.434; T = -4.6; Z = -4.6
Evidence of decreased BMD in the distal radius is often used as an early indicator in hyperparathyroidism. Moreover, tomography and scintigraphy showed osteolytic lesions of the left humeral head and left proximal tibia.
Right inferior parathyroidectomy and right hemithyroidectomy were performed (the second lesion was a thyroid nodule). Parathyroid adenoma with chief cells was confirmed by the histopathological report and the second lesion proved to be a thyroid nodule. After surgery, calcemia and PTH level normalized. Thyroid replacement therapy with 50μg LT4/day was prescribed.
Genetic tests
The HRPT2 and menin genes were studied by amplification of exon and intron regions by PCR and direct sequencing of amplicons. The results were expressed according to the nomenclature of the Human Genome Variation Society.
17 exons were studied for HRPT2 gene and the same mutation was found in all three cases. Nucleotide variation was:
Exon (s) ............................... 6
Nucleotide nomenclature ................ c.505C>T
Proteic nomenclature ................... p. Gln169X
The pathological presence of a stop-codon leads to the synthesis of a truncated protein.
Using Multiplex Ligation-dependent Probe Amplification, the presence of HRPT2 gene deletion or duplication was disproven.
9 exons were studied for MEN1. No nucleotide variations, gene deletion or duplication were found.
DISCUSSION
Compared to sporadic primary hyperparathyroidism, familial hypercalcemia has a lower prevalence, a younger age at diagnosis and an equal ratio between affected men and women. The most frequent histopathological lesion is solitary parathyroid adenoma in sporadic primary hyperparathyroidism and hyperplasia or multiple parathyroid adenomas in familial hypercalcemia (Table 2).
Causes of familial hypercalcemia include hereditary forms of primary hyperparathyroidism and impaired cellular response to extracellular calcium fluctuations.
There are three syndromes associated with primary hyperparathyroidism:
MEN 1 and 2A;
hyperparathyroidism-jaw tumor syndrome (HPT-JT);
familial isolated hyperparathyroidism (FIHP).
Impaired cellular response to extracellular calcium fluctuations is responsible for:
familial benign hypocalciuric hypercalcemia;
severe neonatal hypercalcemia.
MEN1 syndrome has an autosomal dominant inheritance with 100% genetic penetrance by age of 50.2,3 In most cases, primary HPT is the first clinical manifestation.4 Associated tumors are located in the endocrine pancreas, pituitary, adrenal cortex or carcinoid tumors.
The etiology of MEN1 syndrome is menin inactivation. The MEN1 tumor suppressor gene was cloned from 11q13 by positional cloning.5,6 Its product, menin, has been found to bind specifically to JunD, whereas disruption of this binding activity by MEN1 mutations leads to inhibition of JunD-activated transcription.7
Over 100 germline mutations of this tumor suppressor gene have been reported (exons and introns: missense, nonsense, frameshift, ARNm splicing defects). The type of mutation is not associated with a specific phenotype but is present in all affected family members. Genetic testing is required to confirm the clinical diagnosis.2
MEN2A syndrome has an autosomal dominant inheritance with 95% genetic penetrance. Only 25% of these patients have primary hyperparathyroidism.4 Associated tumors are: medullary thyroid carcinoma and feocromocitoma. Etiology of this syndrome is activating mutations of the RET proto-oncogene (cz 10q11.2)8 which encodes a tyrosine kinases receptor. When codon 634 is affected incidence of HPT is maximum.9 RET proto-oncogene mutations have never been documented in patients with familial isolated hyperparathyroidism.
HPT-JT syndrome is an autosomal dominant disorder consisting of parathyroid tumors, ossified jaw fibromas and renal lesions (Wilms tumor, hamartoma, polycystic kidney disease).10 In the parathyroid, potential histopathological lesions are: solitary solid or cystic adenoma, multiple adenomas or carcinoma.11,12
HPT-JT syndrome has an autosomal dominant inheritance with 70% genetic penetrance (smaller if the mutation carrier is a woman).13
Etiology of this syndrome is inactivating mutations of the HRPT2 (CDC73) gene which encodes a tumor suppressor named parafibromin.14-16 Because of the 15-20% of PT carcinoma linked to parafibromin inactivation, HRPT2 is considered to be a marker of parathyroid cancer.
Parafibromin is a protein localized and functionally linked to the nuclear, nucleolar as well as cytoplasmic compartments.17 Parafibromin is considered a tumor suppressor protein based on its ability to induce apoptosis, down-regulate cyclin D1 levels, inhibit G1 to S phase transition as well as to regulate gene expression of various growth factors.18-20 In addition, parafibromin exhibits histone modulating properties, such as recruiting histone methyltransferases and processing histone mRNAs.21-23
Familial benign hypocalciuric hypercalcemia is an autosomal dominant disorder consisting of right-shifted calcium set-point. Over 150 inactivating mutations of the calcium sensing receptor gene (cz 3q13.3-q21) have been reported.24 The heterozygous form associates moderate non-progressive hypercalcemia with hypocalciuria and elevated PTH. The homozygous form, also named severe neonatal hypercalcemia, associates severe hypercalcemia, symptomatic at birth, with very high levels of PTH.25
Familial isolated primary hyperparathyroidism is an autosomal dominant disorder with reduced genetic penetrance. There are two histopathological entities: hyperplasia/multiple parathyroid adenoma and solitary parathyroid adenoma/potential carcinoma.13
FIHP is a genetically heterogenous disease. No specific genetic anomaly has been reported.16 Instead, specific mutations of other forms of familial hypercalcemia have been described.9 That is why FIHP could be considered a mild form with incomplete penetrance or an early stage of the above syndromes.
FIHP with HRPT2 mutations and solitary parathyroid adenoma with a cystic component is considered a variant of HPT-JT syndrome.26,27 It is associated with increased risk for parathyroid carcinoma.28,29
When menin mutation is present, we can find multiglandular involvement but a more favorable clinical progression than in MEN1 syndrome.30 Two separate regions of menin have been shown to separately bind JunD, and at the C-terminus two nuclear localization signals have been identified.7,13 The two reported FIHP-associated MEN1 mutations fall outside all these regions, suggesting a functional basis for this milder variant of MEN1.13,30
Analysis of the previously published MEN1 mutations in FIHP reveals that 41% of these are missense mutations and 37% are frameshift or nonsense mutations.31,33 This contrasts significantly with the situation in patients with MEN1, in whom more than 80% of the germline mutations are nonsense or frameshift and less than 20% are missense mutations.32,33
Five families with isolated HPT and calcium sensing receptor mutations have been reported. Especially mutations in the 3q region in homozygous form can lead to the development of a parathyroid adenoma.3,24,34
An important fact that one must keep in mind is that different mutations of the same gene are responsible for different syndromes (Table 3).
Regarding the pattern of tumor growth, two main types of pathological growth of parathyroid cells have been postulated,3 apparently depending on mutation type.
Most of the sporadic parathyroid adenomas have stopped their proliferation at the time of surgery. This could be explained by a right-shift in set-point for PTH secretion, which is a stimulus for cell growth because of continuous stimulation by the relative hypocalcemia. This clone of cells continues to grow until the secretion of PTH is enough to raise calcemia to a level that matches the new set-point, and then the growth is stopped. Mutations of CaR have been incriminated for the increase in set-point of PTH secretion seen in primary HPT, but it has been difficult to establish the role of CaR in the tumorigenesis of sporadic parathyroid tumors.13
The second pattern of parathyroid growth is considered to be the result of a mutation in a cell cycle gene. In this situation, tumors grow exponentially. Possible genetic events involved are: somatic deletions and amplifications of specific chromosomal regions harboring putative tumor suppressor genes and oncogenes. Overexpression or rearrangement of PTH-cyclinD1/PRAD1 are commonly seen in parathyroid tumorigenesis.35
CONCLUSION
Familial hypercalcemia is a heterogeneous group of disorders. Like other forms of hypercalcemia, it is often asymptomatic, and calcium determination in routine testing is vital for early diagnosis. Complex laboratory investigations confirm the diagnosis and curative treatment is surgical. Genetic testing has an important role in patient evaluation, especially in situations where it is necessary to assess the risk of developing parathyroid carcinoma. Testing of blood relatives requires careful monitoring of those with confirmed mutations.
ACKNOWLEDGMENTS
We acknowledge the Oncogenetics Laboratory of Groupe Hospitalier Cochin, Paris – Pr. E. Clauser, Dr. M. O. North, Dr. N. Hamzaoui for their very good work with the genetic analysis.
REFERENCES
1. Wermers RA, Khosla S, Atkinson EJ, Achenbach SJ, Oberg AL, Grant CS, 2006 Incidence of primary hyperparathyroidism in Rochester, Minnesota, 1993-2001: an update on the changing epidemiology of the disease. J Bone Miner Res 21: 171-177.
2. Brandi ML, Gagel RF, Angeli A, et al, 2001 Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86: 5658-5671.
3. Bilezikian JP, Brandi ML, Rubin M, Silverberg SJ, 2005 Primary hyperparathyroidism: new concepts in clinical, densitometric, and biochemical features. J Int Med 257: 6-17.
4. Gardner DG, Shoback D, 2007. In Greenspan’s Basic and Clinical Endocrinology, Chapter 23, McGraw Hill Medical.
5. Larsson C, Skogseid B, Öberg K, et al, 1988 Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 332: 85-87.
6. Chandrasekharappa SC, Guru SC, Manickam P, et al, 1997 Positional cloning of the gene for multiple endocrine neoplasia-type I. Science 276: 404-407.
7. Agarwal SK, Guru SC, Heppner C, et al, 1999 Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 96: 143-152.
8. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al, 2006 Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Nat Acad Sci USA 103: 15558-15563.
9. Simonds WF, James-Newton LA, Agarwal SK, et al, 2002 Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore) 81: 1-26.
10. Szabo J, Heath B, Hill VM, et al, 1995 Hereditary hyperparathyroidism-jaw-tumor syndrome: the endocrine-tumor gene HRPT2 maps to chromosome 1q21–q31. Am J Hum Genet 56: 944-950.
11. Simonds WF, James-Newton LA, Agarwal SK, et al, 2002 Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore) 81: 1-26.
12. Marx SJ, Simonds SF, Agarwal SK, et al, 2002 Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Min Res 17(suppl. 2): N37-43.
13. Larsson C, 2000 Dissecting the Genetics of Hyperparathyroidism—New Clues from an Old Friend, J Clin Endocrinol Metab 85: 1752-1754.
14. Carpten JD, Robbins CM, Villablanca A, et al, 2002 HRPT2, encoding parafibromin, is mutated in HPT-jaw tumor syndrome. Nat Genet 32: 676-680.
15. Shattuck TM, Valimaki S, Obara T, et al, 2003 Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med 349: 1722-1729.
16. Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ, 2004 Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab 89: 96-102.
17. Bradley KJ, Bowl MR, Williams SE, et al, 2007 Parafibromin is a nuclear protein with a functional monopartite nuclear localization signal. Oncogene 26: 1213–1221.
18. Lin L, Czapiga M, Nini L, Zhang JH, Simonds WF, 2007 Nuclear localization of the parafibromin tumor suppressor protein implicated in the hyperparathyroidism-jaw tumor syndrome enhances its proapoptotic function. Molecular Cancer Research: MCR 5: 183-193.
19. Yang YJ, Han JW, Youn HD, Cho EJ, 2010 The tumor suppressor, parafibromin, mediates histone H3 K9 methylation for cyclin D1 repression. Nucleic Acids Research 38: 382-390.
20. Woodard GE, Lin L, Zhang JH, et al, 2005 Parafibromin, product of the hyperparathyroidism-jaw tumor syndrome gene HRPT2, regulates cyclin D1/PRAD1 expression. Oncogene 24: 1272-1276.
21. Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, et al, 2005 The parafibromin tumor suppressor protein is part of a human Paf1 complex. Mol Cell Biol 25: 612-620.
22. Chaudhary K, Deb S, Moniaux N, Ponnusamy MP, Batra SK, 2007 Human RNA polymerase II-associated factor complex: dysregulation in cancer. Oncogene 26: 7499-7507.
23. Farber LJ, Kort EJ, Wang P, Chen J, Teh BT, 2010 The tumor suppressor parafibromin is required for posttranscriptional processing of histone mRNA. Mol Carcinog 49: 215-223.
24. Pollak MR, Brown EM, Chou YH, et al, 1993 Mutations in the human Ca(2+) sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 75: 1297–1303.
25. Fuleihan GE, Brown EM, Heath HH III 2008 Familial benign hypocalciuric hypercalcemia and neonatal primary hyperparathyroidism. In: Principles of Bone Biology, Bilezikian JP, Raisz LG, Martin TJ, (eds) Academic Press, San Diego; pp, 1327-1345.
26. Teh BT, Farnebo F, Twigg S, et al, 1998 Familial isolated hyperparathyroidism maps to the hyperparathyroidism-jaw tumor locus in 1q21–q32 in a subset of families. J Clin Endocrinol Metab 83: 2114-2120.
27. Cascón A, Huarte-Mendicoa CV, Javier Leandro-García L, et al, 2011 Detection of the first gross CDC73 germline deletion in an HPT-JT syndrome family. Genes Chromosomes Cancer 50: 922-999.
28. Arnold A, 2008 Familial hyperparathyroidism. In: Rosen CJ, (ed) Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, Washington, DC: American Society for Bone and Mineral Research, pp, 361-367.
29. Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP, 2008 Parathyroid Carcinoma. J Bone Miner Res 23: 1869-1880.
30. Kassem M, Kruse TA, Wong F-K, et al, 2000 Familial isolated hyperparathyroidism—a variant of MEN1. J Clin Endocrinol Metab 85: 165-167.
31. Pannett AA, Kennedy AM, Turner JJ, et al, 2003 Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clin Endocrinol (Oxf) 58: 639-646.
32. Thakker RV, 2006 Multiple Endocrine Neoplasia Type 1. In Endocrinology, DeGroot LJ, Jameson JL (eds) Philadelphia, PA: WB Saunders, pp, 3509-3531.
33. Hannan FM, Nesbit AM, Christie PT, et al, 2008 Familial Isolated Primary Hyperparathyroidism Caused by Mutations of the MEN1 Gene. Nat Clin Pract Endocrinol Metab 4: 53-58.
34. Pearce SH, Williamson C, Kifor O, et al, 1996 A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med 335: 1115-1122.
35. Motokura T, Bloom T, Kim HG, et al, 1991 A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature 350: 512-515.
Address for correspondence:
Adina Chemigian, “G.I. Parhon” National Institute of Endocrinology,
Bd. Aviatorilor 34-36, sector 1, Bucharest, 011863,
Tel.: +40 744622743, e-mail: adinaghemi@yahoo.com
Received 17-03-2013, Accepted 10-06-2013