1Department of Endocrinology and Diabetes Center, 2Department of Pathology, 3Department of Radiology, 4Department of Haematology, “G. Genimatas” General Hospital, Athens, Greece
OBJECTIVE: Primary central nervous system (CNS) non-Hodgkin’s lymphoma is a rarely encountered clinical entity. Here we present a case of a primary CNS diffuse large B-cell non-Hodgkin’s lymphoma developed on a previously operated and irradiated pituitary macroadenoma. DESIGN-RESULTS: A 60-year-old woman presented with muscle weakness and eye lid ptosis. Thirty years ago, she was diagnosed with a non-functioning pituitary macroadenoma requiring repeated incomplete operations and conventional radiotherapy and accompanied by partial anterior pituitary deficiency. On admission, the magnetic resonance imaging (MRI) identified a pituitary sellar mass extending into the suprasellar region, compressing the optic chiasm and invading the left cavernous sinus. Following transsphenoidal surgery, the histological investigation revealed the presence of a diffuse large B-cell non-Hodgkin’s lymphoma without other loci from the systemic staging. Following chemotherapy and despite a marked resolution of the neoplastic pituitary mass in the post-chemotherapy MRI scan, the patient’s course was complicated with consciousness deterioration attributed to epileptic seizures and she died of a hospital acquired infection. CONCLUSIONS: Clinicians should include primary CNS lymphoma in the differential diagnosis of an isolated invasive sellar mass. The possible association of primary CNS lymphoma development with the history of operated and irradiated pituitary adenoma is herein discussed.
Irradiation, Non-Hodgkin’s lymphoma, Pituitary lymphoma, Pituitary macroadenoma
INTRODUCTION
Pituitary
adenoma is the most common cause of a sellar mass, accounting for up to 15% of
intracranial neoplasms.1 On the
other hand, primary pituitary lymphomas are very rare, comprising rarely 0.1%
of the cases undergoing transsphenoidal surgery.2 The presentation
of primary pituitary lymphomas can be puzzling since they may be mistaken for
pituitary adenomas.3-5 Only the histological examination can confirm
the diagnosis and determine the designated treatment strategy.
Here we
present an uncommon case of a diffuse large B-cell
non-Hodgkin's lymphoma with primary central nervous system (CNS) involvement.
The lesion developed on a previously operated and irradiated pituitary
macroadenoma. To our knowledge, this is the third case of a primary pituitary
lymphoma developing in patients harbouring a pituitary adenoma and the first to
have arisen following pituitary radiation.6,7 We offer a brief
overview of the existing literature and suggest potential underlying mechanisms
to explain this rare coexistence.
CASE REPORT
A
60-year-old woman presented to our department reporting generalised muscle
weakness and episodes of headache of gradually increasing intensity and
frequency in the last month, as well as right eye
lid ptosis. She is a mother of two healthy children and had a medical history
of a non-functioning pituitary adenoma. Thirty years ago, she had presented with
amenorrhea and vision impairment, mainly in the right eye, and radiologic
evaluation had identified the presence of a macroadenoma with suprasellar
extension; however, data on the precise dimensions are lacking. The hormonal
work-up revealed modest hyperprolactinemia and she underwent a transsphenoidal
adenomectomy; because of incomplete resection of the adenoma, a frontal
craniotomy followed. Unfortunately, the adenoma excision was again incomplete
and the surgery was complicated by left optic nerve damage and sight loss. The
patient developed multiple anterior pituitary deficiencies requiring
hydrocortisone supplementation (20mg daily). The histological investigation
confirmed the presence of a chromophobe pituitary adenoma, but further details
from her medical record are not available. Two years later, and due to acute
right visual loss, she underwent a second, once again incomplete craniotomy and
decompression of the right optic nerve. Afterwards, treatment with conventional
radiotherapy was considered and she received a total dose of 46 Gy resulting in
reduction of the tumour size and improvement of the visual fields. With the
exception of a pituitary magnetic resonance imaging (MRI) two years before
showing no residual adenomatous tissue, the patient's follow-up was not
systematic. The remaining medical and family history was unremarkable.
On
admission, clinical examination revealed third nerve palsy, while biochemical
investigation was normal except for moderate hyponatremia (Na+
124mmol/L). Hormonal work-up confirmed multiple anterior pituitary deficiency:
decreased free thyroxine [8pmol/L, NR 9-21, (0.62ng/dl)] and triiodothyronine
levels [0.90nmol/L, NR 0.89-2.4 (59ng/dl)] with inappropriately normal
thyrotropin levels (TSH 1.2mΙU/L),
suppressed gonadotrophin levels (FSH 2.6IU/L, LH 0.75IU/L), as well as growth
hormone and insulin growth factor 1 levels (GH 0.2 μg/L,
NR 0.6-6.6, IGF-1 42 U/ml, NR 122-327). Morning
cortisol levels [228nmol/L, NR 138-690 (8.26μg/dl)]
were normal (she was under hydrocortisone treatment) and prolactin levels were
24.2ng/ml (NR 1.2-29). There was no evidence of diabetes insipidus.
Unfortunately, measurement of a-subunit levels was not undertaken in our
patient.
Her
symptoms and electrolytic abnormalities resolved with the administration of
intravenous fluids and supplementation with hydrocortisone and thyroxine. A
relapse of the known pituitary macroadenoma was suspected and a pituitary MRI
was performed which identified a pituitary macroadenoma 2.5x2.6cm with
inhomogeneous enhancement following intravenous gadolinium administration,
extending into the suprasellar region, compressing the optic chiasm and
invading the left cavernous sinus and the right side of the pons (Figure 1).
The patient underwent a sublabial transsphenoidal surgery and histological
investigation revealed the presence of neoplastic tissue with intense degenerativealterations
replacing the normal cell architecture. Cell
immunohistochemistry was positive for the leukocyte common antigen (LCA),
whereas the indices for pituitary hormones and epithelial cells were negative.
Moreover, cells were positive to L-26 (CD-20) and PanB, whereas indices to
CD-5, Cyclin-D1, bcl-6, CD10 were negative. Cell proliferation index Ki-67 was
over 30%. 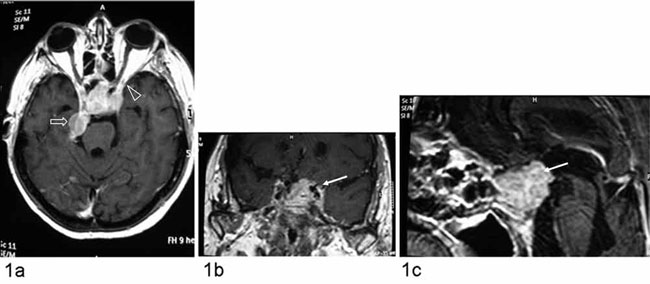
Figure 1. A. Brain MRI scan T1-weighed axial after gadolinium enhancement. B & C. Coronal and sagittal T1-weighed pituitary MRI scan respectively. The pre-treatment figures show a sellar mass with diffuse inhomogeneous enhancement after gadolinium administration, which occupy the intrasellar region, suprasellar cistern, extending and surrounding the left cavernous sinus (→), compressing the optic chiasm and reaching the optic canal (arrowheaad ). The mass produces a slight compression to the pons on the right side in the prepontine cistern (arrow).
The
histological results established the diagnosis of a diffuse large B-cell
non-Hodgkin's lymphoma (NHL) (Figure 2). Evaluation and staging for systemic
disease with thorax, abdomen and pelvis imaging, bone marrow aspiration and CSF
analysis excluded other neoplastic foci. Scanning with Ga-67-citrate and
testing for the HIV were negative. Systemic chemotherapy with intraspinal
methotrexate infusion and the R-MPV chemotherapeutic protocol was
administrated, i.e. rituximab 950mg, methotrexate 6.7g, vincristine 2.6mg and
procarvazine 1.3g cumulative dose. Seven days later, ophthalmoplegic signs
resolved and the post-chemotherapy MRI scan showed a marked regression of the
neoplastic pituitary mass with decompression of optic chiasm and left cavernous
sinus (Figure 3). The patient's course was complicated with an acute decrease
of the consciousness level 20 days after the R-MPV scheme. Her symptoms were
attributed to epileptic seizures and after a long hospitalisation period she
died of a hospital acquired infection. Unfortunately, no autopsy data were
available.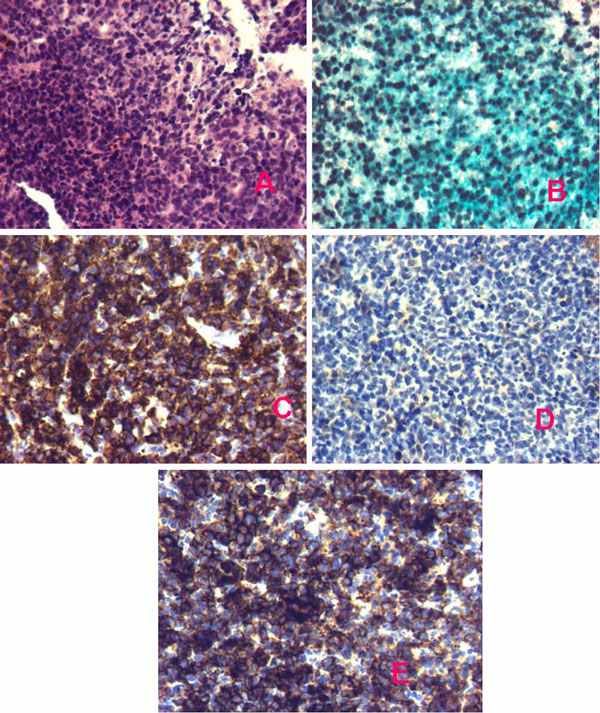
Figure 2. Histochemistry and immunohistochemistry of pituitary neoplastic tissue establishing the diagnosis of a diffuse large B-cell non-Hodgkin’s lymphoma (x20). A. Hematoxylin-eosine staining. B. Ki-67 over 30%. C. L-26 (CD-20). D. PanKer. E. PanB. 
Figure 3. A. Brain T2-weighed axial MRI scan. B & C. Pituitary T1-weighed coronal and sagittal MRI scan respectively after gadolinium enhancement, showing a marked regression of the sellar mass with central area of necrosis (post-chemotherapy).
DISCUSSION
We
describe an unusual case of a primary pituitary lymphoma that developed many
years after surgical and irradiation attempts to treat an invasive pituitary
macroadenoma.
On
presentation, the clinical findings of our patient orientated us to consider a
relapse of the known pituitary adenoma and led to the decision to proceed to a
transphenoidal resection of the residual adenomatous tissue. The unexpected
histological examination altered the initial clinical working hypothesis and,
after exclusion of systemic involvement, the diagnosis of a primary pituitary
B-cell lymphoma in an immunocompetent patient was established. The gradual
onset of her symptoms and the pituitary imaging were not supportive of a
pituitary apoplexy. The hyponatremia was probably related to
adrenocorticotropin and TSH deficiency. Although cortisol levels were within
normal range under replacement therapy, we feel that the patient did not
receive adequate hydrocortisone during this stressful period. Furthermore,
according to her medical records she was not under thyroxine replacement
despite the thyrotroph dysfunction documented on admission. Another factor that
should be mentioned is the inadequate compliance of the patient during therapy
and follow-up.
CNS
non-Hodgkin's lymphoma is associated with a poor prognosis. It usually arises
from metastatic spread of systemic disease, whereas primary CNS
lymphoma (PCNSL) is uncommon, constituting 3% of all
intracranial neoplasms.8-10 Even rarer is the sole involvement of
the hypothalamus- pituitary region.11,12 The majority of cases are
associated with congenital and acquired immunodeficiency, whereas around 29
case reports with sellar and suprasellar location concern immunocompetent
patients, like ours.4,5,13
Regarding
radiologic evaluation, there are no distinct
radiological features of sellar and suprassellar lymphomas that can aid the
differential diagnosis. On MRI, they usually appear as iso- or hypointense on
T1 and T2-weighted images and tend to have homogeneous enhancement following
radio-contrast administration.14
To
our knowledge, only two previous reports have described the simultaneous
existence of a pituitary adenoma with a primary pituitary lymphoma.6,7
Kuhn et al have reported a patient harbouring a mixed T-cell lymphoblastic
lymphoma and a pituitary adenoma with immunoreactivity to FSH, diagnosed 25
years after the initial resection of the adenoma. The second case concerned a
patient with a collision tumour, consisting of pituitary adenomatous tissue
with immunoreactivity to TSH and chromogranin closely admixed with diffuse
large B-cell lymphoma. Our patient had a long history of a recurrent,
previously operated and irradiated macroadenoma, but on histology all pituitary
markers were negative and the pituitary was replaced by
lymphomatous tissue. However, it should be noted that there are null cell
adenomas with negative immunostaining for all pituitary hormones. To the best
of our knowledge, this is the first report of a primary pituitary lymphoma
developing following pituitary radiation for a recurrent macroadenoma.
Brain
radiation for pituitary adenomas is a well established risk factor for second
brain tumour development.15 However, the vast majority of these
malignancies constitute solid tumours, like astrocytomas, meningiomas and
gliomas.16,17 Primary CNS lymphomas are not particularly common in
this setting, even though one could speculate several etiopathogenic pathways.18
One
cannot exclude the possibility that prior radiotherapy for a pituitary adenoma
can exert a carcinogenetic effect on hematopoietic cells, expected to become
clinically evident many years following radiation, not only on the site of
irradiation but also in other CNS sites; however, lymphomas are radiosensitive
and this is contrary to the aforementioned hypothesis. Clearly, the possibility
of a preexisting pituitary lymphoma in our patient is unlikely given the long
history of the pituitary adenomas and the known aggressive behaviour and poor
outcome of these lesions. In addition, an infectious agent, i.e. Ebstein-Barr
or herpes virus, could trigger a chronic lymphocytic inflammation that might
transform to a malignant T-cell proliferation, an attractive hypothesis mostly
in the setting of immunodeficient subjects,19 but not easily
supported in our immunocompetent patient, who after all was proven to have a
diffuse large B-cell lymphoma.
There
is literature data supporting the presence of stem cells in the pituitary.20
These cells, i.e. chromophobes, marginal zone cells, follicular cells,
folliculostellate cells and colony-forming units, have the potential to
transdifferentiate into different cell types. These multipotent cells might
have been activated and proliferated, leading to the development of pituitary
lymphoma. Another possible mechanism could be that pituitary irradiation could
has induced autoantibody formation and this type of hypophysitis might have led
to lymphoma development. This is also supported by evidence showing similar
pathogenetic pathways in primary pituitary lymphoma and lymphocytic
hypophysitis in immunocompetent patients.21 This hypothesis
parallels the development of primary thyroid lymphoma in patients with
Hashimoto thyroiditis. However, the long time period between the initial
adenoma diagnosis and the current presentation of the patient make the scenario
of radiation-induced hypophysitis as the underlying mechanism of lymphoma
development less likely.
Another
possible pathway for lymphoma development on
adenomatous tissues is the growth stimulating effect of various pituitary
hormones. Prolactin, growth hormone, but also gonadotrophins and thyrotropin
have been shown to exert mitogenic effects on normal lymphocytes, as well as
lymphoma cells.22-24 Even though there is a possibility of prior
trophic action of pituitary hormones, the markers of all pituitary hormones on
the present histological examination were negative and the circulating hormonal
levels indicated multiple anterior pituitary deficiencies. Finally, the
expression of specific adhesion molecules on adenomatous cells acting as a
lymphocyte attracting signal could be an alternative pathophysiologic
mechanism, although this might better explain a CNS lymphoma with a different
primary origin invading the sellar area rather than stemming from it.25
In
conclusion, we describe a very rare case of primary aggressive CNS lymphoma
presenting isolated in the pituitary gland, this being to the best of our
knowledge the first case developing many years after radiation therapy of a
pituitary macroadenoma. Although one can provide several arguments to interpret
the development of non-Hodgkin's lymphoma in a previously operated and
irradiated tissue, no definite conclusion can be drawn and most hypothesized
pathophysiologic mechanisms remain speculative. Our report emphasizes the need
for clinical awareness in such perplexing cases which clearly require a
multidisciplinary approach.
Disclosure
summary
The
authors have nothing to disclose.
REFERENCES
1. Gsponer J, De Tribolet N, Déruaz JP, et al, 1999 Diagnosis, treatment, and outcome of pituitary tumors and other abnormal intrasellar masses. Retrospective analysis of 353 patients. Medicine (Baltimore) 78: 236-269.
2. Freda PU, Post KD, 1999 Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am 28: 81-117.
3. Katz BJ, Jones RE, Digre KB, Warner JE, Moore KR, 2003 Panhypopituitarism as an initial manifestation of primary central nervous system non-Hodgkin’s lymphoma. Endocr Pract 9: 296-300.
4. Quintero Wolfe S, Hood B, Barker J, Benveniste RJ, 2009 Primary central nervous system lymphoma mimicking pituitary apoplexy: case report. Pituitary 12: 76-79.
5. Carrasco CA, Rojas-Z D, Chiorino R, González G, 2010 Primary pituitary lymphoma in immunocompetent patient: diagnostic problems and prolonged follow-up. Pituitary 15: 93-96.
6. Kuhn D, Buchfelder M, Brabletz T, Paulus W, 1999 Intrasellar malignant lymphoma developing within pituitary adenoma. Acta Neuropathol 97: 311-316.
7. Au WY, Kwong YL, Shek TW, Leung G, Ooi C, 2000 Diffuse large-cell B-cell lymphoma in a pituitary adenoma: an unusual cause of pituitary apoplexy. Am J Hematol 63: 231-232.
8. Eby NL, Grufferman S, Flannelly CM, Schold SC Jr, Vogel FS, Burger PC, 1988 Increasing incidence of primary brain lymphoma in the US. Cancer 62: 2461-2465.
9. Fine HA, Mayer RJ, 1993 Primary central nervous system lymphoma. Ann Intern Med 119: 1093-1104.
10. Schabet M, 1999 Epidemiology of primary CNS lymphoma. J Neurooncol 43: 199-201.
11. Scully RE, Galdabini JJ, McNeely BU, 1976 Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. N Engl J Med 294: 712-720.
12. Giustina A, Gola M, Doga M, Rosei EA, 2001 Clinical review 136: Primary lymphoma of the pituitary: an emerging clinical entity. J Clin Endocrinol Metab 86: 4567-4575.
13. Li Y, Zhang Y, Xu J, Chen N, 2012 Primary pituitary lymphoma in an immunocompetent patient: a rare clinical entity. J Neurol 259: 297-305.
14. Rao VJ, James RA, Mitra D, 2008 Imaging characteristics of common suprasellar lesions with emphasis on MRI findings. Clin Radiol 63: 939-947.
15. Popovic V, Damjanovic S, Micic D, et al, 1998 Increased incidence of neoplasia in patients with pituitary adenomas. The Pituitary Study Group. Clin Endocrinol (Oxf) 49: 441-445.
16. Brada M, Ford D, Ashley S, et al, 1992 Risk of second brain tumour after conservative surgery and radiotherapy for pituitary adenoma. BMJ 304: 1343-1346.
17. Minniti G, Traish D, Ashley S, Gonsalves A, Brada M, 2005 Risk of second brain tumor after conservative surgery and radiotherapy for pituitary adenoma: update after an additional 10 years. J Clin Endocrinol Metab 90: 800-804.
18. Reni M, Ferreri AJ, Zoldan MC, Villa E, 1997 Primary brain lymphomas in patients with a prior or concomitant malignancy. J Neurooncol 32: 135-142.
19. Paulus W, Jellinger K, Hallas C, Ott G, Müller-Hermelink HK, 1993 Human herpesvirus-6 and Epstein-Barr virus genome in primary cerebral lymphomas. Neurology 43: 1591-1593.
20. Vankelecom H, 2007 Stem cells in the postnatal pituitary? Neuroendocrinology 85: 110-130.
21. Huang YY, Lin SF, Dunn P, Wai YY, Hsueh C, Tsai JC, 2005 Primary pituitary lymphoma presenting as hypophysitis. Endocr J 52: 543-549.
22. Yu-Lee LY, Stevens AM, Hrachovy JA, Schwarz LA, 1990 Prolactin-mediated regulation of gene transcription in lymphocytes. Ann N Y Acad Sci 594: 146-155.
23. Fleming WH, Murphy PR, Murphy LJ, Hatton TW, Matusik RJ, Friesen HG, 1985 Human growth hormone induces and maintains c-myc gene expression in Nb2 lymphoma cells. Endocrinology 117: 2547-2549.
24. Costa O, Bouthet C, Sauvage P, Michel JP, Deschaux P, 1990 Age-dependent LH and FSH effect on the proliferation of women’s peripheral blood lymphocytes in vitro. Int J Immunopharmacol 12: 821-829.
25. Kern WF, Spier CM, Hanneman EH, Miller TP, Matzner M, Grogan TM, 1992 Neural cell adhesion molecule-positive peripheral T-cell lymphoma: a rare variant with a propensity for unusual sites of involvement. Blood 79: 2432-2437.
Address for correspondence:
Labrini Papanastasiou, Department of Endocrinology and Diabetes Center, Athens General Hospital “G. Gennimatas”, 154 Mesogion Avenue 11527,
Tel.:+30 210 7768283, Fax:+30 210 7779146, E-mail: linapapan@yahoo.gr
Received 30-12-11, Revised 12-02-12, Accepted 20-04-12