1Faculty of Medicine, University of Belgrade, 2Clinic of Endocrinology, Clinical Center Serbia, Belgrade, Serbia
CONTEXT: Congenital hypopituitarism is a syndrome which is associated with single or multiple pituitary hormone deficiencies. Mutations in a number of developmental genes have been linked to combined pituitary hormone deficiencies, the most common being mutation in the pituitary homeobox protein prophet of the Pit 1 gene (PROP1). PROP1 exhibits DNA-binding and transcriptional activities. On magnetic resonance imaging, most patients with PROP1 mutation have a hypoplastic pituitary gland. Occasionally, transient pituitary enlargement before definite involution is reported. Kallmann syndrome (KS) is a human developmental genetic disorder which is a clinically (isolated hypogonadotropic hypogonadism-IHH) and genetically heterogeneous disease. Routine neuroimaging in classical IHH is thought to be of limited clinical value and normal anatomy of the hypothalamic-pituitary region is often reported. For neither disorder are there many reports on imaging during adulthood. Nor are there any guidelines concerning long-term imaging follow-up in patients with developmental pituitary disorders. OBJECTIVE: Our aim was to present unusual endocrine and imaging abnormalities which developed in adulthood in two patients with developmental pituitary disorders. CASES: We report a female with combined pituitary hormone deficiencies (GH, TSH, gonadotropin and ACTH), except for prolactin, as a consequence of PROP1 mutation, and a male with KS (anosmia and IHH) due to Kal 2 gene (fibroblast growth factor receptor 1- FGFR1) mutation, both of whom in adulthood presented with prolactinomas. CONCLUSION: Both patients with developmental gene mutations, after long-term correction of their sex steroid status, developed prolactinomas. Although the exact mechanism of pituitary tumorigenesis is not known, we speculate that sex steroids may have facilitated prolactinoma development from the prolactin cell pool which underwent uncontrolled proliferation in the setting of a developmental disorder.
FGFR1 mutation, Hypopituitarism, Hypogonadism, Prolactinoma, PROP1 mutation
INTRODUCTION
A number of genes have been implicated in the etiology of hypopituitarism, in particular mutations in genes encoding for transcription factors involved in pituitary development (Figure 1).1,2 Several transcription factors are involved in the early stages of pituitary development (SOX2, SOX3, HESX1, OTX2, LHX3, LHX4) and abnormalities in these processes are associated with syndrome disorders (midline defects, septo-optic dysplasia, agenesis of septum pellucidum and hypopituitarism). The expression of Prophet of the Pit 1 gene (PROP1) leads to the ontogenesis and differentiation of somatotrophs, thyrotrophs, lactotrophs, gonadotrophs and adrenocorticotrophs.3 PROP1 mutations cause GH, PRL, TSH deficiency in addition to reduced gonadotrophins and ACTH, all resulting from severe reduction of the specific pituitary cell population.4,5 Controversy exists as to the timing and extent of adrenal insufficiency, while it remains unclear whether hypocorticism is a typical feature of PROP1 mutation. Published data show that patients with PROP1 mutation develop at least partial secondary adrenal insufficiency, with a gradual decline of the pituitary adrenal axis function which usually occurs at a mean age of 18 years.6,7 Thus, PROP1 mutation is the most common genetic cause of combined pituitary hormone deficiency (CPHD). The data regarding the pituitary size in humans with PROP1 inactivating mutations are not consistent. On imaging, in most patients with congenital hypopituitarism due to PROP1 mutation, the pituitary gland is hypoplastic, though occasionally transient pituitary enlargement is found.2,5 Voutetakis et al reported an enlarged pituitary gland in five out of 15 patients with PROP1 mutation.8 The same authors have further shown pituitary size fluctuation in a long-term follow-up magnetic resonance imaging (MRI), but this was only until the age of 19 years.9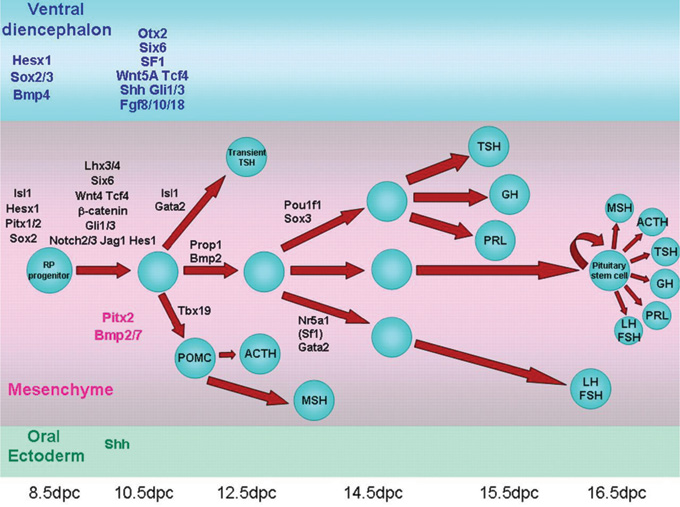
Figure 1. Schematic representation of the developmental cascade of genes implicated in human pituitary development with particular reference to pituitary cell differentiation (reproduced with permission from Kelberman D et al. Endocrine Reviews 2009; 30: 790- 829; Copyright 2009, The Endocrine Society).
Recent investigations into the genetics of isolated hypogonadotropic hypogonadism (IHH) have brought new insights and new questions. Kallman syndrome (KS) is a human developmental genetic disorder characterized by anosmia due to absent olfactory bulbs and IHH. KS is clinically and genetically a heterogeneous disease.10 Different sets of genes have been implicated in KS (KAL1, FGFR1, FGF8, PROKR2, PROK2). Disrupted fibroblast growth factor receptor 1 (FGFR1) signalling has been linked to the etiology of 10% of patients with KS and this gene has been designated as the Kal-2 gene. To date, a significant number of point mutations and deletions of the FGFR1 gene found in KS cases have been reported.11,12 A wide spectrum of olfactory and reproductive phenotypes has been reported.12-14 Patients from the same family sharing an identical mutation may present with classical KS, isolated hypoosmia without hypogonadism and with normosomic IHH. IHH may not be a permanent disorder, as evidenced by reversal to normal activity of the hypothalamic-pituitary-gonadal axis after discontinuation of treatment.15 Routine neuroimaging in classical isolated gonadotropin deficiency is thought to be of limited clinical value and normal anatomy of the hypothalamic-pituitary region is often reported.16
A recently published report suggests the existence of a genetic overlap in KS and CPHD and septo-optic dysplasia.17 Mutations in genes FGFR1, FGF8 and PROKR2 have been found in seven patients with CPHD and septo-optic dysplasia. This suggests a genetic overlap between syndromes affecting the development of the anterior midline in the human forebrain.17 Here we report two patients with developmental hypothalamic-pituitary disorders, a female with congenital hypopituitarism due to PROP1 mutation and a male with KS due to loss of function FGFR1 mutation, both of whom in adulthood presented with peculiar pituitary tumors-prolactinomas.
CASE 1
A 28-year old female with congenital hypopituitarism due to PROP 1 mutation had combined pituitary hormonal deficiencies (GH, gonadotropin, thyrotropin and ACTH deficiencies), except for high levels of prolactin.
She first presented to us at age 19 because of absence of sexual development and with primary amenorrhea. She was born full-term following uncomplicated pregnancy, labor and delivery and grew relatively well. On physical examination she was 166cm tall growing along the 25th percentile as indicated by the growth chart (Figure 2a). She was obese with a body mass index (BMI) 31 kg/m2. We diagnosed congenital hypopituitarism with CPHD (hyposomatotropism, hypogonadism, hypocorticism, hypothyroidism), except for normal prolactin levels. Genetic analysis confirmed a homozygous c.150delA mutation in exon 2 of the PROP1 gene (Pediatric Department, Athens University Medical School, Aghia Sophia Children’s Hospital, Greece and in the Laboratoire de Biologie Moleculaire, Hopital de la Conception, Marseille, France). Pituitary MRI scan was reported normal. Bone mineral density at the lumbar spine was low, Z score was -3.3. For her congenital hypopituitarism she was given replacing regiments of hydrocortisone, L-thyroxine and gonadal steroids except for GH. During the following years she continued growing without GH replacement therapy, reaching a final height of 177 cm at age 22, outgrowing her mother’s height of 170 cm (Table 1, Figures 2a,b; after obtaining informed consent from both the Ethics Committee, the patient and her mother). Bone mineral density improved to Z score equal to – 1 SD. At age 27 she experienced severe headache and irregular menstrual cycles while on sex steroid replacement therapy and her gynecologist requested prolactin measurement. Prolactin levels were high 5554 mU/l, compatible with a prolactinoma. Macroprolactin was excluded.
On MRI (1.5 Tesla) a pituitary microadenoma was reported. Sex steroids were discontinued. Bromocriptine therapy (7.5 mg/day) suppressed prolactin to very low levels. Due to bromocriptine intolerance she was switched to quinagolide. She did not tolerate quinagolide either. Prolactin levels remained high 4950 mU/l and 5200 mU/L. Repeat MRI (3 Tesla) after two years confirmed the presence of pituitary microadenoma with intra-tumoral hemorrhage (Figure 3). A TRH stimulation test was performed at age 30 years and prolactin did not respond to TRH (prolactin during TRH test: 4452, 4812 and 4579 mU/l). Clinical characteristics as well as auxological and hormonal measurement during the 10-year follow-up are shown in Table 1 .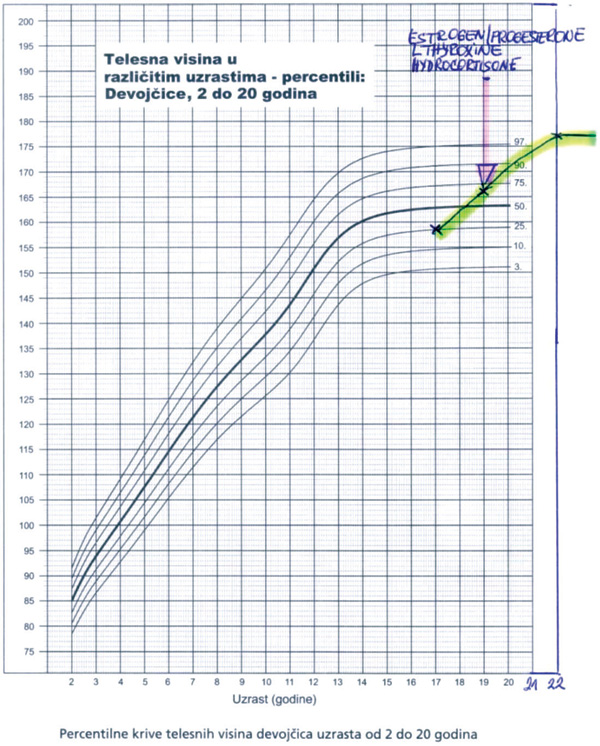

Figure 2. a) Growth chart of patient with congenital hypopituitarism due to PROP1 mutation (growth without growth hormone), b)the patient and her mother.
Figure 3. MRI scan (3 Tesla) of the sellar region of patient with congenital hypopituitarism due to PROP1 mutation who developed microprolactinoma with hemorrhage.
CASE 2
A male patient, 42 years old, presented with severe headache, vomiting, diplopia, hypopituitarism and a large pituitary tumor with apoplexy. He was referred to us for urgent hormonal assessment and replacement prior to surgery. Unexpectedly, past medical history revealed that at age 22 he had been diagnosed with anosmic isolated hypogonadotropic hypogonadism KS. His prolactin level was normal. No cryptorchidism and/or microphallus were reported at birth. Upon successful treatment with GnRH pulsatile therapy for two years he fathered a child at age 25 yrs. In his family history his younger brother also had isolated hypogonadotropic hypogonadism, his father had a history of delayed puberty and his mother had breast cancer. He felt well until age 38 when he developed headache and was treated by an ENT specialist for sinusitis. During the next four years he lost libido and pubic and axillary hair. He was cold intolerant, lost appetite, was tired and reported decreased muscular strength. Suddenly, at age 42, he experienced severe headache and diplopia, felt extremely unwell and began vomiting. Computed tomography of the brain (CT) revealed a large sellar mass, while pituitary MRI scan showed a large intra- and suprasellar mass with tumor apoplexy (Figure 4). The patient was referred to us for urgent correction of hypopituitarism. Hypopituitarism was confirmed except for extremely high levels of prolactin (40.000 mU/L; Table 2 ). We diagnosed a macroprolactinoma with apoplexy and hypopituitarism. Treatment with dopamine agonist bromocriptine (15mg/day) was initiated together with another hormone replacement therapy (hydrocortisone and L-thyroxine). The patient clinically improved and prolactin levels normalized after six months of therapy (Table 2, Figure 5a; after obtaining informed consent from the Ethics Committee and the patient). His hypothalamo-pituitary-adrenal axis recovered, while hypothyroidism, hypogonadism and hyposomatotropism persisted. Testosterone replacement was then introduced. The MRI scan of the pituitary after six months of bromocriptine therapy showed shrinkage of the pituitary macroadenoma (Figure 5b). He remains on L-thyroxine and testosterone replacement therapy. His younger brother with IHH was being treated with testosterone and appears healthy.
Genetic analysis for hypogonadotropic hypogonadism routinely performed in the laboratory of Dr. Tuttelmann (Institute of Human Genetics, Munster, Germany) revealed a novel heterozygous c.570G>A mutation in the fibroblast growth factor receptor-1 (FGFR1) gene in our patient and his brother (both parents died and were not tested for the FGFR1 mutation).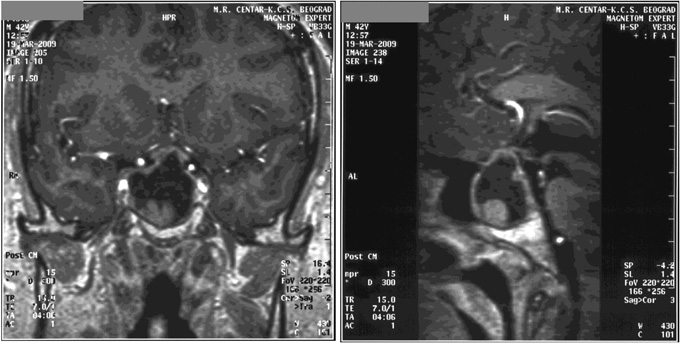
Figure 4. MRI scan of the sellar region of patient with Kallmann syndrome due to FGFR1 gene mutation who developed macroprolactinoma with apoplexy.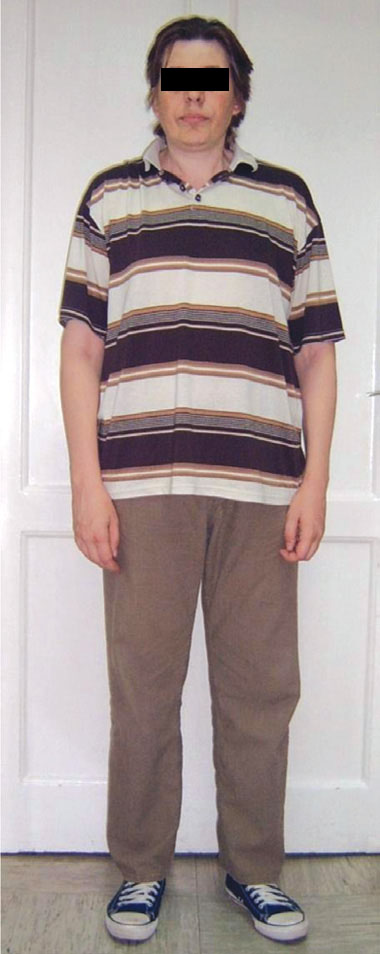

Figure 5. a) The patient, b) MRI scan of the sellar region of patient with Kallmann syndrome due to FGFR1 gene mutation and macroprolactinoma after six months of bromocriptine therapy showing shrinkage of the pituitary macroadenoma.
DISCUSSION
The abovementioned two patients had developmental pituitary gene mutations clinically presented in adulthood with hyperprolactinemia, which was due to the development of prolactinoma. The female patient had congenital hypopituitarism due to PROP1 mutation and developed a microprolactinoma. The male patient had KS due to FGFR1 mutation and developed a macroprolactinoma with apoplexy. Interestingly, the sex-related difference in the size of prolactinomas in our two patients was similar to sporadic prolactinomas in adults, in whom females present more frequently with microprolactinomas, whereas in males macroprolactinomas are much more frequent.18
The case of our female patient with congenital hypopituitarism due to PROP1 is unusual for two reasons. First, despite severe growth hormone deficiency caused by mutation of transcriptional factor PROP1, our patient achieved normal final height (177 cm) and even outgrew her mother. We and others have previously suggested that other factors may be involved in growth, such as insulin, leptin, ghrelin, all of which were elevated in our patient who was obese.19,20 Lazar et al described the growth pattern and final height of five patients with idiopathic CPHD who maintained normal growth despite persistent GH deficiency throughout the growth period.21 These patients did not have hyperprolactinemia or hyperinsulinism. Other GH dependent metabolic parameters in our patient were affected by the lack of GH action. She had increased adipose tissue mass, central obesity and decreased bone mineral density. Similarly, there are reports of the dissociation between the growth-promoting and metabolic effects of GH in patients with “growth without GH” syndrome.22 We hypothesize that our inability to replace her lacking endogenous estrogens during development resulted in delayed epiphyseal fusion, thus facilitating height gain well into adulthood. This is also the case in patients with aromatase deficiency in whom the epiphyseal plate fails to fuse with growth persisting into adulthood.23 Normal height in a PROP1 gene mutation patient with combined pituitary hormone deficiency is rare but has been reported.24,25 Growth without growth hormone is a well-described phenomenon in a number of conditions, which is due to pathology in the hypothalamo-pituitary region, such as in hypothalamic tumors and craniopharyngioma and in patients with septo-optical dysplasia.26
It is of interest that during the 10-year follow-up during which she was on hormone replacement therapy, our patient developed additional endocrine and neuroradiological abnormalities, i.e. hyperprolactinemia and, on MRI, a pituitary mass. To our knowledge, this is the first reported case of a patient with congenital hypopituitarism due to PROP1 mutation who over time developed hyperprolactinemia associated with a possible microprolactinoma. PROP1 transcriptional factor is implicated in differentiation of somatotropes, lactotropes, thyrotropes, corticotropes and gonadotropes and thus is usually associated with low prolactin levels.
However, in some studies normal baseline PRL levels have been reported. Vallette-Kasic et al 2001 reported normal baseline prolactin levels in eight of nine patients with PROP1 mutation, while in other studies normal prolactin levels were less frequent.8,27,28 Lactotroph function may be maintained longer even when other deficiencies develop.6 During the longitudinal follow-up of patients with PROP1 mutations (from 3-33 years) Bottner et al registered progressive decline of anterior pituitary function.6 In our patient, unexpectedly, at age 27 years, after almost ten years of replacement with thyroxine, hydrocortisone and sex steroids, a 10-fold increase in prolactin levels occurred. MRI revealed the presence of a pituitary microadenoma. Pituitary hypoplasia is most frequently observed by MRI; however, various degrees of pituitary enlargement have been seen in one third of patients with PROP1 mutations.29 There are only three reports of patients with PROP1 mutation who were operated on because of the sellar mass lesion.7,29,30 We reported a female at age 22 years with familial PROP1 mutation who presented in adulthood with a large sellar and suprasellar mass.7 Similarly to the report by Parks et al, the histology in our patient revealed an eosinophilic, colloid-like mass and necrotic acellular debris.7,30 In another two operated patients with PROP1 mutation the histology report suggested that pituitary enlargement may be cystic hyperplasia of the intermediate pituitary lobe.29
Voutetakis et al reported pituitary MRI in fifteen patients with PROP1 gene mutations, of whom five patients had pituitary enlargement, and suggested that the pituitary enlargement most likely originated from the intermediate lobe due to absence of physiological regression of the intermediate pituitary lobe during organogenesis.8 However, in our patient the MRI (3 Tesla) did not suggest a cyst-like lesion, which is commonly encountered. The MRI report was compatible with a pituitary microadenoma. MEN I genotyping was negative. Furthermore, after TRH stimulation test, no response of prolactin levels was found, suggesting autonomous prolactin secretion from the pituitary.
Our second patient, with KS, harbored a FGFR1 mutation. His hypogonadism was successfully reversed following hormone replacement treatment for several years but he finally developed a state of emergency because of pituitary tumor apoplexy which was shown to be due to macroprolactinoma. To our knowledge, this has never been reported. In male patients, prolactin secreting tumors occur at a similar frequency throughout the entire lifespan. The majority of functioning pituitary adenomas become symptomatic between the 3rd and 4th decade of life. However, pituitary incidentalomas are clinically nonfunctioning pituitary adenomas, are asymptomatic and are detected as incidental findings on brain magnetic resonance imaging.31 Only a few reports exist describing magnetic resonance imaging of the pituitary in IHH. After analyzing the pituitary by magnetic resonance images in 120 male patients with IHH, Bolu et al found that 18% of patients had incidental pituitary microadenoma.32 They concluded that the incidence of pituitary adenoma in patients with IHH is higher than in the healthy population and that these patients need a close follow-up since these pituitary lesions may play a role in the course of the disease. Our patient with KS came to medical attention for a life-threatening complication (pituitary tumor apoplexy) despite becoming symptomatic several years prior to diagnosis.
Recently it has been reported that mutation in the early developmental transcription factor, such as SOX2, is associated with slowly progressing hypothalamo-pituitary tumor.33 On MRI it appeared as a cystic tumor with suprasellar extension. The nonprogressive nature of these pituitary lesions suggests that they have an embryological origin. Furthermore, Gaston-Massuet et al report craniopharygiomas in mice that overexpress beta catenin in pituitary progenitors during embryogenesis.34 Unlike these tumors that are of embryological origin, our patients developed well differentiated pituitary adenoma-prolactinomas. Molecular mechanisms underlying pituitary tumorigenesis are largely unknown despite a considerable body of information which has been derived from experimental models. One possibility might be the susceptibility to develop tumors in the setting of maldevelopment, similar to the development of testicular tumors in cases with testis dysplasia.35 As for the etiology of prolactinomas, it has been known for many years that chronic treatment with estrogens leads to the development of pituitary tumors in rats and mice and can contribute to the development of prolactinomas in humans.36 It does so via induction of the pituitary tumor transforming factor (PTTG) which has been shown to be tumorigenic by regulating basic fibroblast growth factor (bFGF) secretion.37 Increasing evidence suggests that signals implicated in pituitary development may be relevant to pituitary tumorigenesis, in particular the fibroblast growth factor (FGF) family.37 FGFR2 has a tumor suppressive role. FGFR2 can be epigenetically silenced in the pituitary by estrogen treatment (it enhances histone acetylation and diminishes methylation). Estradiol treatment in experimental conditions induces expression of the normally silent melanoma-associated antigen A3 (MAGE A3) and MAGE A3 is overexpressed in pituitary tumors.38 Thus, epigenetically mediated gene dysregulation is implicated in pituitary tumorigenesis.39,40
Prolactinomas in men tend to exhibit a more aggressive course.18 It has been suggested that although men have lower estrogen levels, as expected, compared to women, their tumors express more estrogen receptors than those in women, this making them more vulnerable to the biological effect of the latter.41 Curiously, our two patients developed prolactinomas long after they had corrected their sex steroid status.
We do not have any clue regarding the possible mechanism involved in their tumorigenesis, but we could tentatively speculate that sex steroids may have facilitated the development of the prolactinoma from the prolactin cell pool which underwent uncontrolled proliferation.
Another possible explanation accounting for the risk in developing pituitary adenoma could be the association with the occurrence of tumors in the families of both patients. The patient with PROP1 mutation had in her family the occurrence of breast cancer, melanoma and lung cancers, while the mother of the patient with KS suffered from breast cancer. The incidence and familial risks in pituitary adenoma and associated tumors has been reported in the Swedish Multigeneration Cancer Registry.42
In conclusion, the adult endocrinologist should be aware that patients with congenital pituitary disorders may acquire a pituitary tumor later in adulthood. There is a need for continual endocrine and, if indicated, neuroradiological monitoring of these patients.
ACKNOWLEDGMENTS
Genetic analysis of the PROP1 gene was performed in the Pediatric Department, Athens University Medical School, Aghia Sophia Children’s Hospital, Greece (Prof. Catherine Dacou) and in the Laboratoire de Biologie Moleculaire, Hopital Moleculaire de la Conception, Marseille, France (Prof. Alain Enjalbert).
Genetic analysis of the FGFR1 gene was performed in the Institute of Human Genetics, University of Munster, Germany (Dr. med. Frank Tuttelmann).
This investigation was supported by a grant from the Ministry of Science of the Republic of Serbia (Project 175033).
The authors have nothing to disclose.
REFERENCES
- Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT, 2009 Genetic regulation of pituitary gland development in human and mouse. Endocrine Rev 30: 790-892.
- Pfaffle R, Klammt J, 2011 Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab 25: 43-60.
- Castinetti F, Davis SW, Brue T, Camper SA, 2011 Pituitary stem cell update and potential implications for treating hypopituitarism. Endocr Rev 32: 453-471.
- Mody S, Brown MR, Parks JS, 2002 The spectrum of hypopituitarism caused by PROP1 mutation Best Pract Res Clin Endocrinol Metab 16: 421-431.
- Kelberman D, Dattani M, 2007 Hypopituitarism oddities: congenital causes. Horm Res 68: Suppl 5: 138-144.
- Bottner A, Keller E, Kratzsch J, et al, 2004 PROP1 mutations cause progressive deterioration of anterior pituitary function including adrenal insufficiency: a longitudinal analysis. J Clin Endocrinol Metab 89: 5256-5265.
- Pekic S, Doknic M, Miljic D, et al, 2011 Case seminar: a young female with acute hyponatremia and a sellar mass. Endocrine 40: 325-331.
- Voutetakis A, Argyropoulou M, Sertedaki A, et al, 2004 Pituitary magnetic resonance imaging in 15 patients with Prop1 gene mutations: pituitary enlargement may originate from the intermediate lobe. J Clin Endocrinol Metab 89: 2200-2206.
- Voutetakis A, Sertedaki A, Livadas S, et al, 2006 Pituitary size fluctuation in long-term MR studies of PROP1 deficient patients: A persistent pathophysiological mechanism? J Endocrinol Invest 29: 462-466.
- Pitteloud N, Durrani S, Raivio T, Sykiotis GP, 2010 Complex genetics in idiopathic hypogonadotropic hypogonadism. Front Horm Re 39: 142-153.
- Kim SH, Hu Y, Cadman S, Bouloux P, 2008 Diversity in fibroblast growth factor receptor 1 regulation: learning from the investigation of Kallmann syndrome. J Neuroendocrinol 20: 141-163.
- Hu Y, Bouloux P, 2010 Novel insights in FGFR1 regulation: lessons from Kallmann syndrome. Trends Endocrinol Metab 21: 385-393.
- Dode C, Levilliers J, Dupont JM, et al, 2003 Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33: 463-465.
- Pitteloud N, Acierno JS Jr, Meysing A, et al, 2006 Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 103: 6281-6286.
- Raivio T, Falardeau J, Dwyer A, et al, 2007 Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 357: 863-873.
- Quinton R, Beirne P, Bouloux P, Stanhope R, Conway G, 2001 Routine neuroimaging in classical isolated gonadotropic deficiency is of limited clinical value. Clin Endocrinol (Oxf) 54: 127-129.
- Raivio T, Avbelj M, McCabe MJ, et al, 2012 Genetic overlap in Kallmann Syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab 97, Feb 8 (Epub ahead of print).
- Delgrange E, Trouillas J, Maiter D, Donchier J, Tourniaire J, 1997 Sex-related difference in the growth of prolactinomas: a clinical and proliferation study. J Clin Endocrinol Metab 82: 2012-2017.
- Doknic M, Pekic S, Djurovic M, Zivkovic V, Popovic V, 2004 Multiple pituitary hormone deficiency (MPHD) associated with normal height, absent puberty and obesity. Pediatr Endocrinol Rev 1: Suppl 3: 505-507.
- Iwayama H, Kamijo T, Ueda N, 2011 Hyperinsulinemia may promote growth without GH in children after resection of suprasellar tumors. Endocrine 40: 130-133.
- Lazar L, Dan S, Phillip M, 2003 Growth without growth hormone: growth pattern and final height of five patients with idiopathic combined pituitary hormone deficiency. Clin Endocrinol (Oxf) 59: 82-88.
- Pavlou M, Tsatsoulis A, Efstathiadou Z, Bitsis S, Papadopoulou ZL, 2001 A study of the growth-promoting and metabolic effects of growth hormone (GH) in a patient with the “growth without GH” syndrome. Growth Horm IGF Res 11: 225-230.
- Rochira V, Zirilli L, Maffei L, et al, 2010 Tall stature without growth hormone: four male patients with aromatase deficiency. J Clin Endocrinol Metab 95: 1626-1633.
- Aroyo A, Permasetti V, Vasilyev V, Amato S, Yen S, Mellon P, 2002 A unique case of combined pituitary hormone deficiency caused by PROP1 gene mutation (R120C) associated with normal height. Clin Endocrinol (Oxf) 57: 283-291.
- Dattani M, 2002 GH deficiency might be associated with normal height in PROP1 deficiency. Clin Endocrinol (Oxf) 57: 157-158.
- Tiulpakov A, Mazerkina N, Brook C, Hindmarsh P, Peterkova V, Gorelyskev S, 1998 Growth in children with craniopharyngioma following surgery. Clin Endocrinol (Oxf) 49: 733-738.
- Vallette-Kasic S, Barlier A, Teinturier C, et al, 2001 PROP1 gene screening in patients with multiple pituitary hormone deficiency reveals two sites of hypermutability and a high incidence of corticotroph deficiency. J Clin Endocrinol Metab 86: 4529-4535.
- Lemos MC, Gomest L, Bastos M, et al, 2006 PROP1 gene analysis in Portuguese patients with combined pituitary hormone deficiency. Clin Endocrinol (Oxf) 65: 479-485.
- Zygmunt-Gorska A, Starzyk J, Adamek D, et al, 2009 Pituitary enlargement in patients with PROP 1gene inactivating mutation represents cystic hyperplasia of the intermediate pituitary lobe. Histopathology and over 10 years follow-up of two patients. J Pediatr Endocrinol Metab 22: 653-660.
- Parks JS, Brown MR, Baumach I, Sanchez JC, Stanley CA, Gianella-Neto D, 1998 Natural history and molecular mechanism of hypopituitarism with large sella turcica. 80th Annual Meeting of the Endocrine Society, 470.
- Freda P, Beckers A, Katznelson L, et al, 2011 Endocrine Society Pituitary incidentaloma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 96: 894-904.
- Bolu S, Tasar M, Uckaya G, Gonul E, Deniz F, Ozdemir IC, 2004 Increased abnormal pituitary findings on magnetic resonance in patients with male hypogonadotropic hypogonadism. J Endocrinol Invest 27: 1029-1033.
- Atazoglou KS, Andoniadou CL, Kelberman D, et al, 2011 SOX2 haploinsufficiency is associated with slow progressing hypothalamo-pituitary tumours. Hum Mutat 32: 1376-1380.
- Gaston-Massuet C, Andoniadou CL, Signore M, et al, 2011 Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci USA 108: 11482-11487.
- Looijenga LH, Gillis AJ, Stoop H, Biermann K, Oosterhuis JW, 2011 Dissecting the molecular pathways of (testicular) germ cell tumour pathogenesis; from initiation to treatment-resistance. Int J Androl 34 (4 Pt 2): e234-51. doi: 10.1111/j.1365-2605.2011.01157.x. Epub 2011 May 12.
- Kovacs K, Stefaneanu L, Ezzat S, Smyth HS, 1994 Prolactin-promoting pituitary adenoma in male-to-female transsexual patient with protracted estrogen administration. A morphologic study. Arch Pathol Lab Med 118: 562-565.
- Heaney AP, Horowitz GA, Wang Z, Singson R, Melmed S, 1999 Early involvement of estrogen induced pituitary tumor transforming gene and fibroblast growth factor expression in prolactinoma pathogenesis. Nat Med 5: 1317-1321.
- Zhu X, ASA SL, Ezzat S, 2008 Fibroblast growth factor 2 and estrogen control the balance of histone 3 modifications targeting MAGE-A3 in pituitary neoplasia. Clin Cancer Res 14: 1984-1996.
- Ezzat S, 2008 Epigenetic control in pituitary tumors. Endocrine J 55: 951-957.
- Dudley KJ, Revill K, Clayton RN, Farrell WE, 2009 Pituitary tumours: all silent on the epigenetics front. J Mol Endocrinol 42: 461-468.
- Stefaneanu L, Kovacs K, Hovarth E, et al, 1994 In situ hybridization study of estrogen receptor messenger ribonucleic acid in human adenohypophysial cells and pituitary adenomas. J Clin Endocrinol Metab 78: 83-88.
- Hemminki K, Forsti A, Ji J, 2007 Incidence and familial risks in pituitary adenoma and associated tumors. Endocrine-Related Cancer 14: 103-109.
Address for correspondence:
Professor Dr Vera Popovic, MD PhD FRCP, Clinic of Endocrinology, Clinical Center Serbia, Serbia, Faculty of Medicine, University of Belgrade, Belgrade, Serbia, Tel.: +381 11 3639702, Fax: +381 11 2685357, e-mail: popver@eunet.rs
Received 11-10-11, Revised 13-12-11, Accepted 17-01-12