1Department of Pediatrics, Division of Endocrinology and Diabetology, University Children’s Hospital, Inselspital, Bern, 2Pediatric Ambulatory Care Centre, Lugano, 3Institute of Pathology, University of Bern, Bern, Switzerland, 4Translational Oncology Research Centre, Dept of Pathology, Queen Alexandra Hospital, Cosham, Portsmouth, UK
Adrenocortical tumors are rare in children and present with variable signs depending on the type of hormone excess. We herein describe the unusual presentation of a child with adrenocortical tumor and introduce the concept of in vitro chemosensitivity testing. CASE REPORT: A 10.5-year-old girl presented with hypertrichosis/hirsutism and weight loss. The weight loss and behavioral problems, associated with halted puberty and growth, led to the initial diagnosis of anorexia nervosa. However, subsequent weight gain but persisting arrest in growth and puberty and the appearance of central fat distribution prompted further evaluation. RESULTS AND FOLLOW-UP: 24h-urine free cortisol was elevated. Morning plasma ACTH was undetectable, while cortisol was elevated and circadian rhythmicity was absent. Thus a hormonally active adrenal cortical tumor (ACT) was suspected. On magnetic resonance imaging (MRI) a unilateral, encapsulated tumor was found which was subsequently removed surgically. Tissue was investigated histologically and for chemosensitivity in primary cell cultures. Although there were some risk factors for malignancy, the tumor was found to be a typical adenoma. Despite this histology, tumor cells survived in culture and were sensitive to cisplatin in combination with gemcitabine or paclitaxel. At surgery, the patient was started on hydrocortisone replacement which was unsuccessfully tapered over 3 months. Full recovery of the hypothalamus-pituitary-adrenal axis occurred only after 3 years. CONCLUSIONS: The diagnosis of a hormonally active adrenocortical tumor is often delayed because of atypical presentation. Cortisol replacement following unilateral tumor excision is mandatory and may be required for months or years. Individualized chemosensitivity studies carried out on primary cultures established from the tumor tissue itself may provide a tool in evaluating the effectiveness of chemotherapeutic drugs in the event that the adrenocortical tumor may prove to be carcinoma.
Adrenal adenoma, Adrenal cortical tumor, Chemosensitivity, Cushing’s syndrome, Steroid withdrawal
INTRODUCTION
Adrenocortical tumors are rare in children. The first case of an adrenocortical tumor in childhood was reported in 1865.1 The worldwide incidence of adrenal tumors in children younger than 15 years ranges from 0.3 to 4 cases per million/per year.2,3 However, incidence shows a broad geographic variation with a high rate in Brazil (3.4-4.3 cases per million/per year) and a low rate in Europe and the USA (0.3-0.38 cases annually per million).2,4 The female to male ratio varies slightly (1.6-1.9:1).5,6 Adrenocortical tumors derive from the cortical cells of the adrenals and may produce steroids autonomously.4,7 In children, unlike in adults, about 90% of all adrenocortical tumors are hormonally active.4
The clinical presentation of hormonally active adrenocortical tumors is related to the type of excess steroid hormones produced.2 Onset is mostly insidious.8 Signs and symptoms of mixed steroid hormone excesses may also be present if tumors secrete more than one type of hormone; however, one type of hormone excess may predominate over time. The variations in clinical manifestations are multiple and often delay diagnosis.2,4
Overall, Cushing’s syndrome is the most common initial clinical manifestation of adrenocortical tumors in children leading to the typical combination of distinct clinical findings, namely rapid weight gain associated with growth retardation or growth arrest.6,9 In children, a significant number of adrenocortical neoplasms presenting with Cushing’s syndrome are malignant, while the opposite is true for adults.8 Treatment of malignant tumors of the adrenal cortex in childhood is still ineffective10 with reported 5-year event-free survival rates of 35-55%.2,5,6
This report aims to raise awareness of the broad clinical spectrum of Cushing’s syndrome in childhood as well as to discuss challenges in diagnosis and management. In addition, the concept is introduced of in vitro chemosensitivity testing as a tool to design drug treatment for highly malignant tumors such as the adrenocortical carcinoma.
CASE REPORT
Initial presentation
A 10.5-year-old girl was examined by a general pediatrician in private practice for hypertrichosis/hirsutism and weight loss (△-2-3 kg). Her weight was 32.2 kg (-0.4 SD), her height was 142.1 cm (0.2 SD) and her BMI was 15.7 kg/m2 (-1.5 SD) (Figure 1). On physical exam she presented with excess lanugo hair on her limbs and on her back. Pubic and axillary hair corresponded to Tanner stage 3.11 She had no breast development and a normal, prepubertal external genitalia. Past medical history was unremarkable except for a post-streptococcal glomerulonephritis diagnosed at the age of 2 years with prolonged hematuria for the subsequent two years. Because of excess hair for age, laboratory investigations were performed (Table 1 ) which revealed no abnormalities other than slightly elevated androstenedione and cholesterol. On follow-up, the girl had a further weight loss of about 3 kg, with weight 29.7 kg (-1.5 SD) and BMI of 14.7 kg/m2 (-1.8 SD). Growth arrest persisted over the following months and she developed behavioral problems which were associated with eating abnormalities. Thus, she was diagnosed with anorexia nervosa and treated with a diet regimen and special psychological counseling. Over the following 5 months the patient gained 4 kg (Figure 1). One year after the initial presentation, she was examined by the general pediatrician who found an arrest in growth and puberty development, a ‘moon face’, central fat distribution and very slim arms and legs with poor muscle mass, and Cushing’s syndrome was suspected. The patient was then referred to our University hospital for further investigations. Repeat laboratory testing revealed undetectable basal ACTH but elevated cortisol and androstenedione values (Table 1). Circadian rhythm of cortisol secretion was absent and 24h urine free cortisol was grossly elevated, confirming the diagnosis of primary hypercortisolism. A high-dose dexamethasone overnight suppression test showed no suppression of morning cortisol (768 nmol/l, cut-off <138). As serum ACTH was suppressed, the patient was diagnosed with ACTH independent cortisol excess. Ultrasound revealed an intraabdominal mass in the upper part of the left kidney. MRI confirmed an adrenal tumor 2.5x2.4x3.2 cm in size on the left side and showed a normal right adrenal (Figure 2). The patient was now 12 years old so that the risk for malignancy of this tumor was uncertain.4 The father of the girl suggested considering chemosensitivity testing of the tumor tissue after surgical removal in a specialized laboratory to search for chemotherapeutic treatment options in the event of malignancy. Unilateral adrenalectomy was performed under pre- and perioperative protection with stress-dose hydrocortisone (100mg/m2) replacement. An encapsulated 9.92 g adrenal tumor was removed (Figure 3). Part of the tumor was sent to Portsmouth for chemosensitivity testing of cultured tumor cells. Histological analysis demonstrated an adrenocortical adenoma (Figure 3). One mitotic figure in 50 HPF, 10% clear cells and absence of atypical mitotic figures, necrosis or capsular invasion resulted in a modified Weiss score of 2.12 The proliferation index using MIB1 staining was 2% (Figure 3). The surrounding non-neoplastic adrenal cortex revealed an atrophy of the zona fasciculata, consistent with hypercorticism (Figure 3).
Figure 1. Growth chart of the reported patient showing height and weight before and after diagnosis and treatment for Cushing’s syndrome.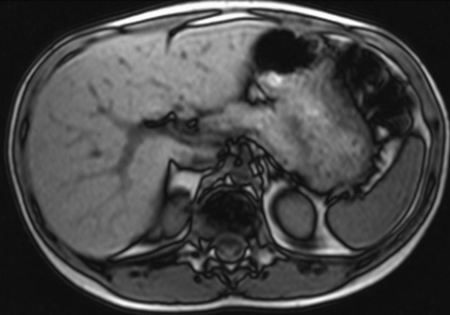
Figure 2. Abdominal MRI showing an adrenal mass above the left kidney.
Figure 3. A: Macroscopic appearance upon surgery. B: Overview with adrenocortical tumor on the right side and atrophic nonneoplastic adrenal cortex missing the zona fasciculata on the left side. C: High power of a field without clear cells and with cytologic atypia, without mitoses. D: Ki-67 staining.
Follow-up
After adrenalectomy, hydrocortisone at a dose of 50mg/m2/d was initiated and slowly tapered over the next 3 months. Nevertheless, the girl developed clinical symptoms of cortisol deficiency manifesting as fatigue and weakness. Low basal serum cortisol was confirmed (Figure 4, Table 1). Therefore, hydrocortisone substitution was reinstalled at a dose of 30mg/m2/d and slowly diminished over the next 18 months. Basal serum ACTH, cortisol and DHEA-S levels were measured at 8 am every 3 months (Figure 4). Over this time period basal ACTH and cortisol levels were noted to normalize and hydrocortisone was discontinued. One month after cessation of hydrocortisone supplementation, a low-dose ACTH test revealed normal basal values but an insufficient rise in cortisol upon stimulation. Thus, the patient was given an emergency pass for steroid substitution in situations of stress such as fever, severe illness or operations. Three years after tumor extirpation, metyrapone and low-dose ACTH tests showed normal response in both tests. Thus steroid substitution in stress situations was suspended.
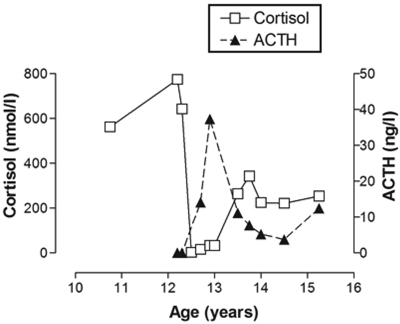
Figure 4. Plasma cortisol and ACTH profile from diagnosis to final testing.
METHODS
Biochemical testing
To characterize the form of hypercortisolism (pituitary versus adrenal disease) in our patient, we performed a high (single)-dose 8 mg/1.73m2 dexamethasone overnight suppression test as described.8 By contrast, we used the low-dose (500ng/1.73m2 iv) ACTH test and the overnight metyrapone test (30mg/kg;) to assess adrenal recovery after tumor surgery.13
Establishment of primary cultures of the adrenal tumor and chemosensitivity studies
Chemosensitivity testing was performed using the standardized ATP-based chemosensitivity assay (ATP-TCA) as previously described.14 This involved setting up a short-term primary cell culture from the biopsy material and exposing the growing cells to single agents and combinations of anticancer drugs for 6 days, following which the inhibition (% of control, untreated cells) at all 6 concentrations of each agent or combination tested was obtained by the luciferin-luciferase assay.14
DISCUSSION
The challenge to diagnose Cushing’s syndrome
In many patients the onset of Cushing’s is insidious8 and the clinical manifestations variable. In our patient initial presentation with hypertrichosis, weight loss and behavioral problems led to the erroneous diagnosis of eating disorder. This diagnosis was only reconsidered when subsequent weight gain was not followed by improved growth and pubertal development as well as signs of Cushing’s syndrome. In children, leading signs of Cushing’s are rapid weight gain (92%) and growth arrest (84%) followed by osteopenia, fatigue, arterial hypertension and delayed or arrested puberty.15 By contrast, essential obesity in children is characterized by a continuous weight gain which is accompanied by excessive growth. In our patient the initial laboratory work-up for hirsutism revealed a high-normal cortisol and a slightly elevated androstenedione and cholesterol. Clearly, these findings are not sufficiently specific to suspect the diagnosis. To diagnose hypercortisolism, the first two screening tests are serum or salivary cortisol profile measurements to assess circadian rhythm and/or 24 hours urine free cortisol.16,17 While early morning basal serum/salivary cortisol levels may remain in the normal range in patients with cortisol-producing tumors, cortisol levels often remain high in tumor patients during the day differing clearly from the tightly regulated circadian rhythm of cortisol production that reaches a nadir around midnight.18-21 If clinical suspicion for hypercortisolism is high and screening tests are negative, one should insist and repeat the screening tests. In androgen producing adrenal tumors, one may find grossly elevated levels of the normally produced adrenal androgens DHEA/-S and androstenedione; but such tumors may also produce the more potent androgen testosterone, which is not normally produced in the human adrenal cortex.3,22 Depending on the hormonal production of the adrenal tumor as well as the responsiveness/resistance of the individual system to these hormones, patients will present with very subtle to severe signs of hormonal excess, this accounting for the delay in the diagnosis of the disease in many patients, including ours.
The challenge to avoid adrenal crisis after hypercortisolism
At the time of diagnosis of Cushing’s syndrome when our patient presented with characteristic symptoms, urinary free cortisol (UFC) secretion rate increased more than 10-fold to 1547 nmol/24h (normal range 10-40). This corresponds to a prednisone therapy of about 1 mg/kg/d, which is a recommended dose for the treatment of inflammatory or immunologic disorders in pediatrics.
Supraphysiologic glucocorticoid exposure, exogenous or endogenous, bears the risk of shutting down the hypothalamic-pituitary adrenal axis completely over a period of time. Thus, it is common practice to taper GC therapy if used at supraphysiologic doses for more than 2-4 weeks; similar tapering schemes are found in all endocrine textbooks.23,24 When removing the adrenal tumor in patients with Cushing’s syndrome, the patient is at great risk for Addison’s crisis because of the sudden lack of endogenous cortisol and the increased need to respond to the acute stress of anesthesia and surgery. Therefore, patients with CS need stress-dose cortisol replacement before being taken to the operating room. Thereafter, they need cortisol replacement therapy and emergency stress-dose adjustments lifelong if bilateral adrenalectomy was performed. With unilateral adrenalectomy, replacement therapy is mandatory until recovery of the HPA axis is confirmed. In our patient, we started hydrocortisone replacement therapy at a dose of 50mg/m2/d post surgery and tapered the dose over 3 months. Nevertheless, she developed symptoms of adrenal insufficiency, prompting us to restart replacement and taper dose at a much lower pace over the next 18 months. Finally, 21 months after tumor removal, her HPA axis had recovered fully based on clinical presentation and biochemical testing. In fact, although Addison’s crisis is a rare event, it occurs most often after GC dose reduction or owing to lack of stress-related dose adjustments.24 It is not always easy to anticipate the optimal duration and dosing scheme of steroid substitution, especially after Cushing’s syndrome, due to tumors of unknown onset and rate of cortisol production. Certainly, recovery time was initially underestimated in our patient. Recovery of the HPA axis and resumption of normal cortisol production by the contra-lateral adrenal cortex after unilateral adrenalectomy may take up to 3 years.4,25
Adrenal tumors of the cortex and their treatment
Adrenocortical tumors are mostly benign. Cushing’s syndrome due to benign adrenal tumors may be caused by adenomas or by bilateral hyperplasia histologically manifesting as primary (non-)pigmented, micro- or macronodular disease.26 For a comprehensive overview of the different forms of benign adrenal hyperplasias, their histopathologic as well as genetic characteristics and the genetic syndromes associated, we refer the reader to detailed reviews by Stratakis.26-29
At any age, adrenal Cushing’s syndrome is most often caused by a unilateral adenoma, although in about 10% bilateral adenomas may be found.30,31 Malignant tumors are rare both in adults and children32,33 Characteristics associated with an increased risk for malignancy include female sex, young/old age (<5/>80 years), unilateral tumor and tumor size >5-7 cm.2,4,26,34 Other risk factors for an adverse outcome include urinary excretion of 17-OHCS of >4 mg/m2/d, age older than 3.5 years, an interval of more than 6 months between first signs and diagnosis and virilization at clinical diagnosis.2,4,32 However, the most important single risk factor seems to be tumor size. Thus, our patient presented with some risk factors for potential malignancy of her adrenal tumor.
The most successful treatment of adrenocortical tumors (ACT) is surgery. However, until the end of the 1940s, most children undergoing adrenalectomy for a hormone-secreting ACT died postoperatively due to acute adrenocortical crisis. Only when cortisone became widely available did the outcome improve dramatically. Today, unilateral as well as bilateral adrenalectomy is mostly performed laparascopically with good postoperative outcome. Nevertheless, longterm outcome of malignant tumors, especially with metastatic disease or incomplete tumor resection, is poor.2,4,5 In addition, heterogeneity and rarity of the disease make it very difficult to establish prognostic factors and evaluate adjuvant treatment modalities. To date, mitotane (o,p’-DDD) is the most widely studied drug used adjuvant to surgical treatment or in inoperable patients.35,36 In patients with progression of disease despite mitotane and surgery, cytotoxic therapy may be added. However, chemotherapy resistance is an issue, as adrenocortical tissue produces high levels of the multi-drug resistance protein MDR1 (P-glycoprotein) which functions as a drug efflux pump.37 So far, the best response rate achieved in adult patients has been reported with etoposide, doxorubicin and cisplatin in combination with mitotane.38 Although the efficacy of chemotherapeutic agents as single or combination therapies in pediatric patients was uncertain,6,10,39,40 newer results involving 254 cases from the International Pediatric Adrenocortical Tumor Registry suggest a role for chemotherapeutic agents in the treatment of patients with advanced disease.5 Novel drugs directed against targets altered in adrenocortical carcinomas such as epidermal growth factor receptor (EGFR; ErbB-1; HER1), Insulin-like Growth Factor 1 (IGF-1) Receptor, Vascular endothelial growth factor ( VEGF) are currently under investigation in ongoing randomized international trials in adults.37,38,41
Chemosensitivity studies of primary tumor cell cultures
To our knowledge, our patient is the first pediatric ACT to undergo chemosensitivity testing. Establishing primary cell cultures from malignant tumor tissues to test for specific drug responsiveness in vitro is a potentially useful approach, especially when dealing with rare tumors for which no optimized treatment protocols exist. The role of chemotherapy in the management of childhood ACT has not been fully established,6,8,36,38,40 although some tumors are responsive to mitotane- or cisplatin-containing regimens like EDP (Etoposide, Doxorubicin and Cisplatin).5
In our patient, the scientist father had some experience with chemosensitivity testing of tumors in vitro and discussed with us the possibility of sending tumor tissue for performance of chemosensitivity studies to intervene in the event that we were faced with an adrenocortical carcinoma. Thus primary cultures were established and drug cocktails were designed and tested on the basis of published literature.4,31,34,36,42
The results (Figure 5) showed cisplatin to be the most effective single agent tested, though combinations including cisplatin were more inhibitory than cisplatin alone and cisplatin with gemcitabine might have been a potentially useful treatment option if the tumor had proved to be malignant. Chemosensitivity testing using the ATP-TCA depends on the survival of tumor cells in a serum-free culture medium with or without drugs and is not a test for malignancy. Accordingly, in our case the ATP-TCA revealed results, although the histological investigations showed that the tumor was neoplastic but unlikely to be malignant.
Figure 5. ATP-based tumor chemosensitivity assay. Inhibition of cell survival (%) in serum-free medium for (A) single agents and (B) combinations tested at six dilutions. Cisplatin is the most active single agent tested and cisplatin combinations show the greatest activity of those tested. The 100% test drug concentrations were as follows: 5-FU - 45 μg/ml, cisplatin - 3.80 μg/ml, docetaxel - μg/ml, doxorubicin x3 - 1.5 μg/ml, gemcitabine - 12.5 μg/ml, paclitaxel - 6.8 μg/ml, and vinorelbine 12 μg/ml. For combinations, drugs were added at these concentrations and diluted together from 200 - 6.25% in the assay.
In summary, patients with Cushing’s syndrome often present with atypical and subtle symptoms. Therefore, if CS is clinically suspected, it should be searched via several biochemical screening tests and, if necessary, on more than one occasion. In pediatric patients most adrenal tumors are benign. Malignant adrenal tumors are rare and have a poor prognosis. Surgery is the treatment of choice for adrenal tumors, while chemotherapeutic options are scarce. Chemosensivity studies using primary cultures established from tumor tissue may provide an additional option when searching for effective drugs against individual cancers.
ACKNOWLEDGEMENTS
We thank the patient and her family for their willingness to share their medical history and results with the scientific community. We also thank Prof. Roland Ammann and Prof. Kurt Leibundgut, University Children’s Hospital Bern, for helpful discussion.
REFERENCES
1. Anonymous 1865 General melasma and short hair over the entire body of a child of three years, with conversion of the left supra-renal capsule into large malignant tumor; external organs of generation resembling of adult life. Lancet 1: 175.
2. Sandrini R, Ribeiro RC, DeLacerda L, 1997 Childhood adrenocortical tumors. J Clin Endocrinol Metab 82: 2027-2031.
3. Wolthers OD, Cameron FJSI, Honour JW, Hindmarsh PC, Savage MO, Stanhope RGBCGD, 1999 Androgen secreting adrenocortical tumours. Arch Dis Child 80: 46-50.
4. Ribeiro RC, Michalkiewicz EL, Figueiredo BC, et al, 2000 Adrenocortical tumors in children. Braz J Med Biol Res 33: 1225-1234.
5. Michalkiewicz E, Sandrini R, Figueiredo B, et al, 2004 Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 22: 838-45.
6. Hanna AM, Pham TH, Askegard-Giesmann JR, et al, 2008 Outcome of adrenocortical tumors in children. J Pediatr Surg 43: 843-849.
7. Visser HK, 1966 The adrenal cortex in childhood. 2. Pathological aspects. Arch Dis Child 41: 113-136.
8. Stratakis CA, 2008 Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin- independent Cushing syndrome). Endocr Dev 13: 117-132.
9. Magiakou MA, Mastorakos G, Oldfield EH, et al, 1994 Cushing’s syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med 331: 629-636.
10. Ciftci AO, Senocak ME, Tanyel FC, Buyukpamukcu N, 2001 Adrenocortical tumors in children. J Pediatr Surg 36: 549-554.
11. Marshall WA, Tanner JM, 1969 Variations in pattern of pubertal changes in girls. Arch Dis Child 44: 291-303.
12. Aubert S, Wacrenier A, Leroy X, et al, 2002 Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol 26: 1612-1619.
13. Miller WA, Achermann JC, Flück CE, 2008 The adrenal cortex and its disorders. Pediatric Endocrinology 3rd (ed) chapter 12.
14. Cree IA, 1998 Luminescence-based cell viability testing. Methods Mol Biol 102: 169-177.
15. Devoe DJ, Miller WL, Conte FA, et al, 1997 Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab 1997: 3196-3202.
16. Nieman LK, Biller BM, Findling JW, et al, 2008 The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 93: 1526-1540.
17. Arnaldi G, Angeli A, Atkinson AB, et al, 2003 Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 88: 5593-5602.
18. Knutsson U, Dahlgren J, Marcus C, et al, 1997 Circadian cortisol rhythms in healthy boys and girls: relationship with age, growth, body composition, and pubertal development. J Clin Endocrinol Metab 82: 536-540.
19. Debono M, Ghobadi C, Rostami-Hodjegan A, et al, 2009 Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab 94: 1548-1554.
20. Frisch H, Parth K, Schober E, Swoboda W, 1981 Circadian patterns of plasma cortisol, 17-hydroxyprogesterone, and testosterone in congenital adrenal hyperplasia. Arch Dis Child 56: 208-213.
21. Krieger DT, Allen W, Rizzo F, Krieger HP, 1971 Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab 32: 266-284.
22. Lee PD, Winter RJ, Green OC, 1985 Virilizing adrenocortical tumors in childhood: eight cases and a review of the literature. Pediatrics 76: 437-444.
23. Stewart PM 2003 The adrenal cortex. Philadelphia: WB. Saunders Company, 2002.
24. Arlt W, Allolio B, 2003 Adrenal insufficiency. Lancet 361: 1881-1893.
25. Klose M, Jorgensen K, Kristensen LO, 2004 Characteristics of recovery of adrenocortical function after treatment for Cushing’s syndrome due to pituitary or adrenal adenomas. Clin Endocrinol (Oxf) 61: 394-399.
26. Stratakis CA, 2009 New genes and/or molecular pathways associated with adrenal hyperplasias and related adrenocortical tumors. Mol Cell Endocrinol 300: 152-157.
27. Horvath A, Boikos S, Giatzakis C, et al, 2006 A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet 38: 794-800.
28. Bourdeau I, 2004 Clinical and molecular genetic studies of bilateral adrenal hyperplasias. Endocr Res 30: 575-583.
29. Horvath A, Giatzakis C, Robinson-White A, et al, 2006 Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res 66: 11571-11575.
30. Stratakis CA, Boikos SA, 2007 Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab 3: 748-757.
31. Latronico AC, Chrousos GP, 1997 Extensive personal experience: adrenocortical tumors. J Clin Endocrinol Metab 82: 1317-1324.
32. Allolio B, Hahner S, Weismann D, Fassnacht M, 2004 Management of adrenocortical carcinoma. Clin Endocrinol (Oxf) 60: 273-287.
33. Libe R, Fratticci A, Bertherat J, 2007 Adrenocortical cancer: pathophysiology and clinical management. Endocr Relat Cancer 14: 13-28.
34. Bugg MF, Ribeiro RC, Roberson PK, et al, 1994 Correlation of pathologic features with clinical outcome in pediatric adrenocortical neoplasia. A study of a Brazilian population. Brazilian Group for Treatment of Childhood Adrenocortical Tumors. Am J Clin Pathol 101: 625-629.
35. Terzolo M, Angeli A, Fassnacht M, et al, 2007 Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 356: 2372-2380.
36. Schteingart DE, 2007 Adjuvant mitotane therapy of adrenal cancer - use and controversy. N Engl J Med 356: 2415-2418.
37. Berruti A, Ferrero A, Sperone P, et al, 2008 Emerging drugs for adrenocortical carcinoma. Expert Opin Emerg Drugs 13: 497-509.
38. Fassnacht M, Allolio B, 2009 Clinical management of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab 23: 273-289.
39. Rescorla FJ, 2006 Malignant adrenal tumors. Semin Pediatr Surg 15: 48-56.
40. Teinturier C, Pauchard MS, Brugieres L, Landais P, Chaussain JL, Bougneres PF, 1999 Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol 32: 106-111.
41. Fassnacht M, Kreissl MC, Weismann D, Allolio B, 2009 New targets and therapeutic approaches for endocrine malignancies. Pharmacol Ther 123: 117-141.
42. Wooten MD, King DK, 1993 Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer 72: 3145-3155.
Address for correspondence:
Christa E. Flück, M.D., Pediatric Endocrinology and Diabetology,
Univ Children’s Hospital Bern, Freiburgstrasse 15 / G3 812,
3010 Bern, Switzerland, Tel.: +41 31 632 0499, Fax: +41 31 632 8424,
E-mail: christa.flueck@dkf.unibe.ch
Received 30-01-11, Revised 20-02-11, Accepted 10-13-11