1Department of Endocrinology, 2Radiology, G. Genimatas General Hospital, Athens Greece; 39th Pulmonary Department, "SOTIRIA" Hospital, Athens, Greece; 4Unit of Dermatology, Royal Postgraduate Medical School, London, UK
We describe a 57-year old female with the diagnosis of skull and meningeal Langerhans' cell histiocytosis who was treated with the combination of azathioprine and methotrexate. Although the skull lesions improved considerably on this regimen, the patient developed diabetes insipidus while the anterior pituitary function remained intact.
Langerhans cell histiocytosis, Diabetes insipidus, Skull, Meninges, Bone, Pituitary, Azathioprine, Methotrexate
INTRODUCTION
Langerhans' Cell Histiocytosis (LCH) is a rare disease of variable biologic behavior, resulting from the pathologic proliferation of cells with morphologic characteristics of Langerhans cells originally described in the epidermis1. The course of the disease is fairly unpredictable as it may spontaneously resolve or progress to a disseminating stage, compromising vital functions with severe or even fatal consequences1,2. Although LCH was originally reported as a non-specific infiltrating disease, biopsies from adults' and children's lesions, with either single or multisystem involvement, showed proliferation of a single Langerhans cell clone2,3.
The spectrum of the clinical manifestations of the disease varies and virtually any organ can be affected4. LCH can be stratified according to system involvement as either single system disease (single site: single bone lesion, isolated skin disease, solitary lymph node, or multiple site: multiple bone lesions, multiple lymph node involvement), or multisystem disease (multiple organ disease, with or without dysfunction)5. The Hypothalamic-Pituitary system (HPS) is involved in 5-50% of cases, with most patients presenting with diabetes insipidus (DI)4,6-9. Anterior pituitary dysfunction has been described in up to 20% of patients usually associated with DI8,10-14. However, anterior pituitary function has not been systematically studied in adults and most clinical information has been obtained from studies in children7,11,13,15. In this report, we describe an adult female patient with LCH, who initially presented with multiple site bone disease but subsequently developed DI despite improvement of the bony lesions. The endocrine manifestations of LCH, its clinical course and response to treatment are discussed.
PATIENT REPORT
A 57-year old woman presented with a 9-month history of right scalp pain and a subsequent nodular lesion in the right parietal bone. She was initially treated with local steroids but, due to the persistence of the lesion, she underwent local excision; histology was consistent with a non-specific inflammatory lesion. Two months later the lesion re-appeared; in addition, there was loss of the integrity of the right parietal bone (3 cm in diameter) and a further lesion 6-7 cm in diameter was found laterally extending into the occipital scalp (Figure 1). There were no other local or systemic complaints. A further local excision of the primary and satellite lesions was performed. Histological examination was consistent with LCH based on the presence of characteristic Langerhans cells with prominent nuclear groove exhibiting positive immunohistochemistry to S-100 protein and CD1α antibody.

Figure 1. X-Ray film profile examination of calvarium. There are three lytic lesions with sharp edges located in the cortex of parietal and temporo-occipital bones.
Endocrine manifestations of LCH
Radiological examination at presentation revealed an infiltrating lesion, 2x3 cm in diameter, at the apex of the right parietal bone, and an additional 1.5x2 cm lesion at the same side. There were no abnormal findings in the examination of the skin, respiratory, cardiovascular, abdominal, neurological and musculoskeletal system. Examination of the urogenital system was normal. Laboratory investigations at that time were within normal limits except for slightly raised C-reactive protein levels [15.2 mg/l (0-5 mg/l)]. Radiological skeletal survey showed no additional abnormal findings. Endocrine investigation revealed normal anterior and posterior pituitary function (Table); lung function tests were normal. Magnetic Resonance Imaging (MRI) of the brain and pituitary showed dural infiltration extending into the parietal and frontal region (Figure 2a), and extra-cranial extension involving the periosteum and calvarium (parietal and frontal). In addition, further soft tissue masses were found in the left cerebellopontine angle and at the planum sphenoedale; the pituitary fossa was partially empty (Figure 2b). The patient was considered to have localized LCH of the skull and treatment with azathioprine 50 mg three times daily and methotrexate 5 mg once weekly was initiated. Two weeks later methotrexate was increased to 10 mg. Follow-up of the patient one and two months later while on therapy revealed persistently elevated CRP levels. The skull lesions changed in number and size due to peripheral sclerosis of the initial infiltrations (Figure 3), suggesting initiation of a healing process5.


Figure 2a. MRI - T1 Weighted axial image with administration of intravenous contrast medium indicating a dural infiltration in the frontal and parietal region extending to the periosteum and calvarium.

Figure 2b. MRI - T1 Weighted coronal image with administration of intravenous contrast medium. There is a prolapse of suprasellar cistern into the pituitary fossa, image of partially empty sella. The infundibulum is in the midline and the pituitary gland is slightly compressed down to the floor of pituitary fossa.
Three months later the patient developed polyuria, nocturia and polydipsia. Initial assessment revealed normal Plasma/Urine osmolalities and serum electrolytes. However, two weeks later serum sodium increased to 154 mEq/L (135-145 mEq/L) while serum and urine osmolality were 327 and 300 mOsm/kg, respectively, following a water deprivation test. The diagnosis of central DI was made and the patient was started on treatment with oral DDAVP with resolution of her symptoms and normalization of serum and urine osmolalities. Further evaluation of the anterior pituitary function revealed no other hormonal deficiency. Radiological re-evaluation of the skull lesions showed some deterioration (Figure 4), while MRI of the pituitary revealed thickening of the pituitary stalk (not present on the original MRI) (Figure 5). Following this development, the methotrexate was increased to 20mg per week and two and five months later (6 and 9 months after treatment initiation, respectively) the radiological re-evaluation of the skull lesions revealed great improvement with reduction of the size of the lesion and considerable peripheral sclerosis (Figure 6); no other endocrine deficiency was found.
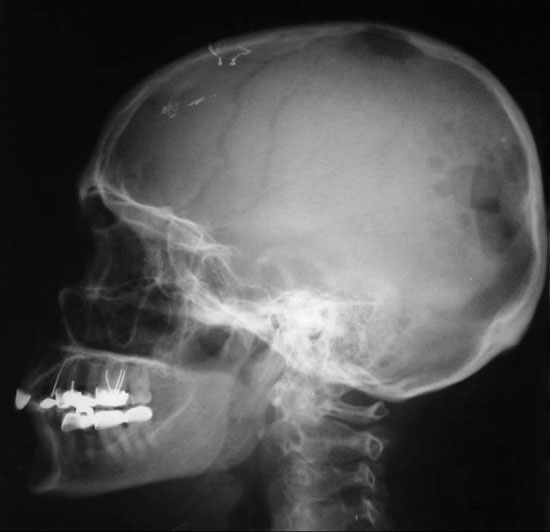
Figure 3. X-Ray film taken two months after initiation of treatment showing the proliferation of the lesions in the temporo-occipital bones.

Figure 4. X-Ray film taken four months after initiation of treatment, showing notable increase of the number of the lesions in the temporo-occipital as well as in the parietal bones.
DISCUSSION
LCH is a rare disease mostly encountered in paediatric patients, with the peak of age range estimated at 1-3 years and an incidence of 3-5 cases per million16. LCH in adults may present at any age and is usually misdiagnosed or under-diagnosed, either because of mild and common clinical symptoms or due to lack of specific symptoms, thus making it difficult to estimate its exact incidence16. In large single institution series, adult LCH comprised about 30% of all cases9. Until prospective studies are performed, an incidence rate in the adult population of about 1 in every 560,000 could be cautiously proposed17.

Figure 5. MRI - T1 Weighted coronal image with administration of intravenous contrast medium showing remarkable thickening of the pituitary stalk resulting in slight compression of optic chiasm.

Figure 6. X-Ray film taken nine months after initiation of treatment. The skull lesions decreased in number and size; a peripheral sclerosis is also noted suggesting the healing process.
Multisystem disease with skull vault defects, and notably temporal bone or orbital lesions, is usually associated with an almost threefold increased risk for developing DI, an increased risk for central nervous system infiltration and anterior pituitary involvement with concomitant hormonal deficits4,6,8. Skull lesions are mainly lytic and may extend intracranially or impinge on the dura18. Anterior pituitary dysfunction in patients with LCH has almost always been associated with DI6,15,10-12,19; only a few cases of pituitary hormone insufficiency without DI have been noted in the literature20. Although DI may predate the diagnosis of LCH, it develops most commonly at about 12 months after the diagnosis is made6,7,15,21.
This pattern of development was also obvious in our patient who initially presented with skull lesions and subsequently developed DI. Although at the time of the latest assessment no further hormonal deficit was encountered, this patient should be at regular follow-up and assessment of anterior pituitary function as deficiencies have been described in a considerable number of patients with DI that had adequate follow-up6. Growth hormone deficiency (GHD) is usually the first endocrine defect observed in addition to DI, with a median latency of about 1 year from diagnosis4,22. GHD occurs in approximately 40% of affected children and has been related to histiocytic infiltration of the hypothalamus22,23. As adults with GHD may show an increase in well-being and a favourable metabolic profile in response to GH therapy, assessment of GHD may be an important part of the evaluation of adult patients with LCH6. There are only a few reports on gonadal function in patients with LCH demonstrating abnormalities of gonadotropin secretion, particularly cases of amenorrhea in adults6. Similarly, thyroid hormone deficiency can be a major component of anterior pituitary dysfunction in patients with LCH6, whereas ACTH deficiency is mostly found in the context of generalized pituitary involvement. However, a case of isolated ACTH deficiency has also been described15.
Efforts to identify predictors of late endocrine sequealae in children with LCH concluded that dynamic endocrine pituitary testing was not a useful predictor. Neither the site of involvement nor the extent of the disease was associated with further endocrine deterioration23. Therefore, it seems that only DI in association with abnormal HPS imaging identifies patients with LCH at higher risk for anterior pituitary dysfunction6. DI with structural changes in the HPS often heralds the involvement of LCH in other parts of the brain with more global neurological or neuropsychologicala sequealae, depending on the location of the involvement8,24.
It has been suggested that none of the therapeutic regimens currently available is able to alter the course of LCH other than stabilisation of the disease25. Moreover, established endocrine abnormalities seem to be permanent10,25. LCH-I and LCH-II studies in children have established some general treatment guidelines; the selection of treatment chiefly depends on the extent of the disease, which must always be evaluated carefully after a systemic diagnostic approach26. In cases of endocrine involvement, systemic chemotherapy appears to be of little benefit in controlling the progression of the disease over a long term, while focal radiotherapy may halt local disease progression in terms of mass effects6. Radiation therapy can also be used in vertebral lesions or lesions of the femoral neck with a risk of fracture or collapse. Other treatments that could be used systemically in the future include: anti-TNF agents which are currently being tested, bone marrow transplantation, and anti-CD1a which was initially used in the diagnostic evaluation but is also promising as a possible therapeutic approach5. In our patient the development of DI is in accordance with the high prevalence of posterior pituitary dysfunction in patients with LCH and skull lesions. Although established adult treatment protocols do not exist, the recommended treatment for patients with a similar clinical presentation, according to the LCH-III protocol, would have been either velban-prednisone-6-merkaptopurine or this 3-drug combination with methotrexate18. Our patient opted for the combination of azathioprine and methotrexate, which was well tolerated. Despite a substantial initial improvement of the skull lesions, the patient developed DI during the course of therapy. Hence, established DI or DI occurring during therapy or persisting does not constitute a predictive sign of therapeutic failure. Additionally, the improvement of skull lesions could either be the result of treatment or could also reflect a spontaneous remission commonly seen during the course of the disease4.
The increasing awareness of the presence of LCH as well as the rising number of investigative procedures and therapeutic options for patients with LCH suggests that their management is best dealt with by a multidisciplinary team. Such a team may include physicians with a special interest in LCH and systems of particular involvement and specialists such as radiologists and oncologists. All available investigative procedures should be reviewed and a consensus on the best-evidence-based management should be agreed upon. As adults with LCH are rare, optimum management should be performed in centers with relevant experience and expertise in order to evaluate the results of current treatment, establish guidelines and develop new therapeutic trials27.
In summary, adult patients with LCH skull lesions are at high risk for the development of DI. The persistence of DI during therapy does not predict failure of the therapeutic modality applied. Although established adult treatment protocols do not exist, therapeutic guidelines emerging from children's studies and treatment protocols could be applied in adult patients.
REFERENCES
1. Schmitz L, Favara BE, 1998 Nosology and pathology of Langerhans Cell Histiocytosis In: Egeler RM, D'Angio GJ, (eds) Hematology / Oncology Clinics of North America (Langerhans Cell Histiocytosis), Saunders, USA; 12: 221-246.
2. Yu RC, Chu C, Buluwela L, Chu AC, 1994 Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet Mar 26: 767-768.
3. Willman CL, Busque L, Griffith BB, et al, 1994 Langerhans'-cell Histiocytosis (histiocytosis X)--a clonal proliferative disease. N Engl J Med 331: 154-160.
4. Arico M, Egeler R 1998 Clinical aspects of Langerhans cell histiocytosis. In: Egeler RM, D'Angio GJ, (eds) Hematology/Oncology Clinics of North America (Langerhans Cell Histiocytosis), Saunders, USA 12: 247-258.
5. Broadbent V, Gadner H 1998 Current therapy for Langerhans Cell Histiocytosis In: Egeler RM, D'Angio GJ, (eds) Hematology/Oncology Clinics of North America (Langerhans Cell Histiocytosis), Saunders, USA; 12: 327-338.
6. Kaltsas GA, Powles TB, Evanson J, et al, 2000 Hypothalamo-Pituitary abnormalities in adult patients with Langerhans Cell Histiocytosis: Clinical, endocrinological, and radiological features and response to treatment. J Clin Endocrinol Metab 85: 1370-1376.
7. Dunger DB, Broadbent V, Yeoman E, et al, 1989 The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med 321: 1157-1162.
8. Grois N, Favara B, Mostbeck H, Prayer D 1998 Central nervous disease in Langerhans cell histiocytosis In: Egeler RM, D'Angio GJ, (eds) Hematology/Oncology Clinics of North America (Langerhans Cell Histiocytosis), Saunders, USA; 12: 327-338.
9. Malpas J 1998 Langerhans Cell Histiocytosis in adults In: Egeler RM, D'Angio GJ, (eds) Hematology/Oncology Clinics of North America (Langerhans Cell Histiocytosis), Saunders, USA; 12: 259-268.
10. Braunstein GD, Kohler PO, 1972 Pituitary function in Hand-Schuller Christian disease. N Engl J Med 286: 1225-1229.
11. French Langerhans Cell Histiocytosis Group 1996 A multicentric retrospective survey of Langerhans' cell histiocytosis: 348 cases observed between 1983 and 1993. Arch Dis Child 75: 17-24.
12. Willis B, Ablin A, Weinberg V, Zoger S, WaraWM, Matthay KK, 1996 Disease course and late sequelae of Langerhans cell histiocytosis: 25-year experience at the University of California, San Francisco. J Clin Oncol 14: 2073-2082.
13. Hanna CE, LaFranchi SH, 1983 Evolving hypopituitarism in children with central nervous system lesions. Pediatrics 72: 65-70.
14. Ahmed M 1993 Endocrine abnormalities in patients with Langerhans cell histiocytosis [Abstract]. Proc of the 75th Annual Meet of The Endocrine Soc.; 73.
15. Broadbent V, Dunger D, Yeomans E, Kendall B, 1993 Anterior pituitary function and computed tomography/magnetic resonance imaging in patients with Langerhans cell histiocytosis and diabetes insipidus. Med Pediatr Oncol 21: 649-654.
16. Broadbent V, Egeler RM, Nesbit ME, 1994 Langerhans Cell Histiocytosis - clinical and epidemiological aspects. Br J Cancer 70: Suppl XXIII: 11-16.
17. Carstensen H, Ornvold K, 1993 The epidemiology of Langerhans Cell Histiocytosis in children in Denmark 1975-89. Med Pediatr Oncol 21: 387-388.
18. McClain KI 2003 Langerhans Cell Histiocytosis (histiocytosis X, eosinophilic granuloma) In: Rose BD (ed), USA; 11: 1-15.
19. Malpas JS, Norton AJ, 1996 Langerhans cell histiocytosis in the adult. Med Pediatr Oncol 27: 540-546.
20. Tabarin A, Corcuff JB, Dautheribes M, et al, 1991 Histiocytosis X of the hypothalamus. J Endocrinol Invest 14: 139-145.
21. Maghnie M, Villa A, Arico M, et al, 1992 Correlation between magnetic resonance imaging of posterior pituitary and neurohypophyseal function in children with diabetes insipidus. J Clin Endocrinol Metab 74: 795-800.
22. Dean HJ, Bishop A, Winter JSD, 1986 Growth hormone deficiency in patients with histiocytosis X. J Pediatr 109: 615-618.
23. Maghnie M, Bossi G, Klersy C, Cosi G, Genovese E, Arico M, 1998 Dynamic endocrine testing and magnetic resonance imaging in the long term follow-up of childhood Langerhans cell histiocytosis. J Clin Endocrinol Metab 83: 3089-3094.
24. Gadner H, Heitger A, Grois N, Gatterer-Menz I, Ladisch S, 1994 Treatment strategy for disseminated Langerhans cell histiocytosis. DAL-HX 83 study. Med Pediatr Oncol 23: 72-80.
25. Kilpatrick SE, Wenger DE, Gilchrist GS, Shives TC, Wollan PC, Unni KK, 1995 Langerhans' cell histiocytosis (histiocytosis X) of bone. Cancer 76: 2471-2484.
26. Gadner H, Grois N, Arico M, et al, 2001 A randomised trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr 138: 728-734.
27. Makras P, Kaltsas G, Samara C, et al, 2003 The spectrum of endocrine abnormalities in patients with Langerhans Cell Histiocytosis (LCH). [Abstract]. Proc of the 7th Annual Meeting of the Neuroendocrinology Section of the German Society of Endocrinology (DGE). Experimental and Clinical Endocrinology & Diabetes 111: 385.
Address correspondence and requests for reprints to:
Dr Gregory Kaltsas, Department of Endocrinology
G. Genimatas Hospital, Athens, Greece,
Tel:+302107796043, Fax:+30 210 7779146, e-mail: gkaltsas@acci.gr
Received 06-11-03, Revised 05-12-03, Accepted 15-12-03