1Department of Surgery, 2Department of Pathology, “Hygeia” Hospital, Athens, Greece
Objective: Adrenal tumors present with clinical features and signs unique to their specific hormonal hypersecretion. However, there have been cases in which the clinical expression has been in conflict with the histologic features of the tumor. In this communication we report an unusual clinical presentation of an adrenal cortical tumor with histologic features of an oncocytoma that clinically mimicked a pheochromocytoma. Design: A 49-year old man was referred to our Unit due to type B aortic dissection and a mass of the left adrenal gland. Computed tomography and magnetic resonance imaging confirmed the presence of aortic dissection extending from the left subclavian artery to both iliac arteries and also revealed a 6 cm tumor on the left adrenal gland. Preoperative endocrine evaluation showed a near tenfold increase of urinary vanillylmandelic acid (VMA) and metanephrine values. Results: Transperitoneal laparoscopic adrenalectomy was successfully performed. The adrenal tumor proved to be an adrenal cortical neoplasm with histologic features of oncocytoma. Conclusion: Although the case of an adrenal cortical adenoma clinically mimicking a pheochromocytoma has been described in the literature, to the best of our knowledge, there has been no previous report of an adrenal cortical neoplasm with predominant features of oncocytoma.
Adrenal tumor, Aortic dissection, Laparoscopic adrenalectomy, Oncocytoma, Pheochromocytoma
INTRODUCTION
Adrenal tumors present with clinical features and signs unique to their specific hormonal hypersecretion. Nevertheless, there have been certain cases in which the clinical expression has been in conflict with the histologic features of the tumor. In the present report we describe a laparoscopic adrenalectomy performed for a tumor with clinical and laboratory findings consistent with pheochromocytoma, which, however, had histopathologic features of an adrenal cortical neoplasm with predominant features of oncocytoma.
CASE REPORT
A 49-year old man was referred to our Unit due to type B aortic dissection and a left adrenal mass. He had a long history (>30 years) of heavy smoking and a history of cocaine abuse. Hypertension was reported, but never treated. The rise of the blood pressure was described as paroxysmal and was associated with episodes of diaphoresis and palpitations. Hypoglycemic attacks or orthostatic hypotension were not mentioned.
Forty days prior to admission, the patient suffered a severe thoracic pain of abrupt onset that led to urgent admission to another hospital. Computed tomography (CT) and magnetic resonance imaging (MRI) showed an aortic dissection extending from the left subclavian artery to both iliac arteries, as well as a 6 cm left adrenal mass (Figure 1). At the time of urgent admission, renal function was seriously impaired and necessitated hemodialysis (serum creatinine 12.3mg/ dl and blood urea nitrogen 250mg/dl).
A detailed laboratory endocrine evaluation revealed markedly increased 24 hr urine vanillylmandelic acid (VMA) and metanephrine levels [522 μmol/d (normal range: 5-55 μmol/d) and 5.98 mmol/mol creatinine (normal range: 0.03-0.74 mmol/mol creatinine), respectively]. Urine catecholamine levels were also elevated [epinephrine: 346 nmol/d (normal range: 0-109.3 nmol/d) and norepinephrine: 842.4 nmol/d (normal range: 82.8-473 nmol/d)]. Blood and 24 hr urine cortisol levels were normal [34.2 nmol/d (nor mal range: 17.1-53.5 nmol/d), 125.2 nmol/d (normal range: 55.1-193.1 nmol/d), respectively] and plasma adrenocorticotropin hormone (ACTH) levels were also normal [5.5 pmol/l (normal range 1.98-11.4 pmol/l)]. Serum potassium was 4.7 mmol/l (normal range: 3.5-5.1 mmol/l), serum aldosterone was 12.4 nmol/l (normal range: 8.3-44.3 nmol/l), renin was 0.33 pmol/l (normal range: 0.12-0.9 pmol/l) and aldoste-rone to renin ratio was 1.10 ng/ml/h (normal range: 0.5-4.7 ng/ml/h). Dehydroepiandrosterone sulphate (DHEA-S) and testosterone blood levels were within normal limits. There were no clinical signs of Cushing's syndrome. Assessment of cardiovascular status with ultrasound and arterial coronography revealed left ventricular dilatation, ejection fraction ~25%, but no significant major coronary artery stenosis.
With the presumptive diagnosis of pheochromocytoma associated with a type B aortic dissection the patient received typical preparation regimen. This consisted of intravenous administration of normal saline (N/S) 0.9% and oral phenoxybenzamine in increasing doses for one week. Subsequently, a left laparoscopic adrenalectomy through transabdominal approach was performed. With the patient in the left lateral decubitus position, the operative technique attained included establishment of the pneumoperitoneum at the umbilicus followed by insertion of two 10 mm trocars at the midclavicular and anterior axillary lines and a 5 mm trocar at the middle axillary line. Despite the technical and hemodynamic challenges posed by the presence of the dissecting aneurysm, the laparoscopic adrenalectomy was completed successfully without any intraoperative hazards.
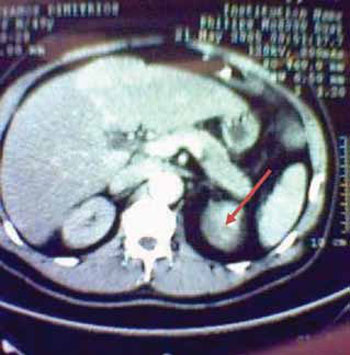
Figure 1. Presence of a 6 cm left adrenal mass (arrow).
The patient had an uneventful hospital stay and was discharged on the third postoperative day in excellent condition. His blood pressure levels were normal in the early postoperative period without any paroxysmal attack and the renal function was normal. During a follow-up period of 12, 24 and 36 months, he has had no hypertensive attacks and no signs of tumor recurrence. 24hr urinary VMA and metanephrine levels have been found within normal range throughout his follow-up period [4.3 mg/24h (normal range: 2.6-7.7) and 0.46 μg/mg creatinine (normal range: 0.05-1.2 μg/mg creatinine): 36 months postoperatively].
Pathology report
The surgical specimen weighed 120 gr and consisted of a 9.5 x 8 x 5 cm adrenal gland mostly occupied by a circumscribed, solid and soft to elastic orange or tan colored tumor measuring 6 x 5 x 4cm.
On light microscopy, H&E sections showed a circumscribed, encapsulated neoplasm consisting of trabecular and alveolar nests of cells. The nests were surrounded by a prominent thin-walled vascular network. The neoplastic cells were large and polygonal with ample, intensely eoshinophilic granular cytoplasm, round slightly irregular nuclei and prominent nucleoli ("oncocytic appearance", Figure 2). Focally, the neoplastic cells were similar to those of a zona fasciculata of the adrenal cortex. Mitotic activity, necroses or capsular/vascular invasion were not evident. The tumor stroma was focally infiltrated by lymphocytes. The residual adrenal cortex showed nodular hyperplasia.
Immunohistochemical stains were performed with the NEXES® automatic immunostainer (VENTANA Medical Systems, Tucson, Arizona, USA). The neo-plastic cells were positive for Synaptophysin and focally positive for pankeratin AE1/AE3, calretinin and BCL2. Less than 1% of neoplastic cells were positive for p53 and Ki67. No positivity was observed for Chromogranin (Figure 3), S100, CD 57 and cyto-keratins 8, 18, 19 (CAM5.2). The immunophenotype was similar to that of the adjacent adrenal cortex.
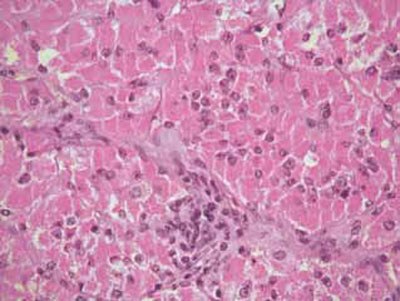
Figure 2. Alveolar nests of large, polygonal neoplastic cells with abundant, granular, eosinophilic cytoplasm, separated by thin fibrovascular septae. Nuclei of neoplastic cells are round and show no significant atypia (H&Ex400, original magnification). 
Figure 3. Neoplastic cells are immunohistochemically negative for Chromogranin (x400, original magnification).
It was concluded that the morphology and im-munohistochemistry were consistent with an adrenal cortical neoplasm with predominant features of adrenal oncocytoma. According to the morphologic parameters of the Weiss system, considered to be predictive of the biologic behavior of conventional (non-oncocytic) adrenocortical tumors, this tumor is classified as borderline or tumor of uncertain malignant behavior [1 minor criterion positive due to the weight of the specimen (>100gr) and the size of the tumor (>5cm)].
DISCUSSION
Adrenal tumors present with clinical features and signs unique to their specific hormonal hypersecretion. Nevertheless, there have been a small number of cases in which the clinical expression has been in conflict with the pathologic features of the tumor. In this study we report a laparoscopic adrenalectomy performed for a tumor with clinical and laboratory findings consistent with pheochromocytoma which, however, had histo-pathologic features of an adrenal cortical neoplasm with predominant features of oncocytoma.
Coexistence of cortical and medullary features in adrenal cells has been recognized since 1960 and these cells have been termed "mixed" or "corticomedul-lary".1 The presence of cortical and medullary features within the same neoplastic cells of adrenal tumors has been repeatedly reported in the literature and the bipotential expression of these tumors has been based on both immunohistochemical and electron microscopy studies.2,3
Adrenal cortical oncocytomas are rare tumors which, in their vast majority, are incidentally discovered non-secreting neoplasms.4,5 However, exceptions to this rule may occur: for example, in a case reported by Xiao et al, an adrenal cortical oncocytoma presented as Cushing syndrome in a 53yr old female patient.6 In another case reported by Erlandson et al, there was raised urinary 17-ketosteroids in a female patient with virilization who subsequently returned to normal after resection of an oncocytic cortical adenoma.7 Geramizadeh et al8 reported another case of secreting adrenal oncocytoma. Although the occurrence of an adrenal cortical adenoma clinically mimicking a pheochromocytoma has been reported in the literature,3 to the best of our knowledge there has been no previous report of an adrenal cortical neoplasm with predominant features of oncocytoma.
Certainly, our case with the characteristic clinical signs and laboratory findings of pheochromocytoma was rather puzzling. Since the histologic features supported the diagnosis of adrenal oncocytoma, the presence of increased cathecholamine secretion made the consideration of other tumor types as sources of the catecholamines quite plausible. Adrenomedul-lary hyperplasia, ganglioneuromas, extra-adrenal paragangliomas, chemodectomas, medullary thyroid carcinomas or carcinoids are potentially sources of increased catecholamines preoperatively. Clinical and imaging studies failed to detect the presence of any extra-adrenal tumor. Well-established clinical guidelines dictate no need for metaiodobenzyl-guanidine (MIBG) scanning in cases of sporadic pheochromocytoma; since we had a documented adrenal mass and a relevant clinical presentation, we preferred to proceed to surgery without MIBG scanning preoperatively. The complete resolution of hypertensive attacks in our patient as well as the normalization of the urinary VMA and metanephrines values throughout the follow-up period of 36 months should be considered confirmatory, namely that the adrenal tumor was the source of the catecholamine hypersecretion. Thus, on clinical grounds, no indication for MIBG scanning in the postoperative follow-up period was considered necessary.
The histological and immunohistochemical findings in our case were consistent with those in previously published cases of adrenal oncocytoma.9-11 Absence of immunohistochemical expression of Chromogranin A along with architectural and cytological features, including the presence of focal adrenal adenoma features, helped in differentiating this tumor from a pheochromocytoma. Immunohistochemical expression of Synaptophysin, a neuroendocrine marker, is well known to be present in both adrenocortical tumors and pheochromocytomas.6
In summary, this communication describes an extremely rare case of adrenal oncocytoma with a clinical presentation mimicking pheochromocytoma and associated with increased values of VMA and catecholamines, findings which posed a considerable diagnostic dilemma pre-operatively.
REFERENCES
1. Kovacs K, Horvath E, 1973 Ultrastructural features of corticomedullary cells in a human adrenocortical adenoma and in a rat adrenal cortex. Anat Anz 134: 387-393.
2. Alsabeh R, Mazoujian G, Goates J, Medeiros LJ, Weiss LM, 1995 Adrenal cortical tumors clinically mimicking pheochromocytoma. Am J Clin Pathol 104: 382-390.
3. Ivsic T, Komorowski R, Sudakoff G, Wilson S, Datta M, 2002 Adrenal cortical adenoma with adrenalin-type neurosecretory granules clinically mimicking a pheo-chromocytoma. Arch Pathol Lab Med 126: 1530-1533.
4. Sang Yong Song, Sunhoo Park, Seong Rim Kim, Yeon-Lim Suh, 2004 Oncocytic adrenocortical carcinomas: A pathological and immunohistochemical study of four cases in comparison with conventional adrenocortical carcinomas. Path Intern 54: 603-610.
5. Sasano H, Jang WF Jr, Chrousos GP, Koch CA, Giordano TJ, Kawashima A 2004 Adrenal cortical adenoma. In DeLellis RA, Lloyd RV, Heitz PU, Eng C (eds), World Health Organization Classification of Tumors, Pathology & Genetics, Tumors of Endocrine Organs, IARR Press, Lyon: p, 145.
6. Xiao GQ, Pertsemlidis DS, Unger PD, 2005 Functioning adrenocortical oncocytoma: a case report and review of the literature. Ann Diagn Pathol 9: 295-297.
7. Erlandson RA, Reuter VE, 1991 Oncocytic adrenal cortical adenoma. Ultrastruct Pathol 15: 549-546.
8. Geramizadeh B, Norouzzadeh B, Bolandparvaz S, Sefid-bakht S, 2008 Functioning adrenocortical oncocytoma: a case report and review of literature. Indian J Pathol Microbiol 2008 51: 237-239.
9. Kitching PA, Patel V, Harach HR, 1999 Adrenocortical oncocytoma. J Clin Pathol 52: 151-153.
10. Sasano H, Suzuki T, Sano T, et al, 1991 Adrenocorti-cal oncocytoma-a true non-functioning adrenocortical tumor. Am J Surg Pathol 15: 949-956.
11. Lin B, Bonsib S, Mieran G, et al, 1998 Oncocytic adrenocortical neoplasm. A i m J Surg Pathol 22: 603-614.
Address for correspondence:
Linos D. MD, Harvard Medical School, Director of 1st Surgical Clinic, "Hygeia" Hospital, 7 Fragoklisias Street, Marousi 151 25, Athens, Greece, Tel.: 01130-2106125001, Fax: 01130-2106126170, E-mail: dlinos@hms.harvard.edu
Received 25-01-10, Revised 04-08-10, Accepted 12-11-10