Receptor Biology Section, Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
Endocrine Disrupting Chemical,Estrogens, Diethylstilbestrol, Genistein, Lavender Oil, Methoxyacetic Acid, Short-Chain Fatty Acid, Tea Tree Oil
INTRODUCTION
A large number of natural and synthetic chemicals have been identified that are reported to disrupt the normal functioning of the endocrine system and produce untoward effects in hormone-responsive target tissues and organs in both humans and animals. These chemicals are broadly referred to as endocrine disrupting chemicals (EDCs) and consist of numerous physically and chemically distinct compounds. The physicochemical differences among EDCs are paralleled by unique biological effects and mechanisms of action within this class of compounds. The purpose of this review is to provide an overview of the physiological effects and unique mechanisms of action of four distinct EDCs that alter estrogen signaling. Two of these compounds, diethylstilbestrol (DES) and genistein (GEN), have been extensively studied and serve as classic examples of EDCs, while the other two compounds, methoxyacetic acid (MAA) and the essential oils of lavender and tea tree, have only recently been described as EDCs.
DIETHYLSTILBESTROL
Diethylstilbestrol (DES) is a nonsteroidal synthetic estrogen developed by Sir Charles Dodds and colleagues in 1938.1 During the late 1940’s physicians began using DES to treat pregnant women in an attempt to maintain normal placental steroid levels and to prevent miscarriages and premature births.2 It was not until 1953 that a randomized controlled clinical trial evaluating the effectiveness of DES in preventing miscarriage and premature birth was reported.3 Results from this study showed that DES offered no protective effects with respect to miscarriage or premature birth. Despite the outcome of this clinical trial, DES continued to be prescribed to pregnant women until 1971, when it was reported that in utero DES exposure was associated with a rare reproductive tract cancer, termed vaginal clear cell adenocarcinoma, in approximately 0.1% of daughters of women who had taken the drug.4,5 As a result of this association the FDA advised against using DES in pregnant women in 1971. Subsequent studies have reported multiple adverse effects in males and females as a result of prenatal DES exposure that occur with greater frequency than clear cell adenocarcinoma. In females, these include anatomical malformations of the cervix, vagina, and uterus, decreased fertility, and fewer successful pregnancies.6-9 In contrast to females, no increased incidence of cancer has been reported in males prenatally exposed to DES. However, adverse effects have been observed in their reproductive tracts, including testicular hypoplasia, cryptorchidism, and epididymal cysts.6,7,10
Epidemiological analyses continue for those men and women exposed to DES in utero as physicians and scientists strive to better understand the long-term effects in humans. These efforts have been accompanied by intensive laboratory-based research to elucidate the mechanisms by which DES elicits its carcinogenic and teratogenic effects and to better predict and prevent the untoward effects of in utero DES exposure in humans. Toward this end, a variety of rodent models have been developed to study the mechanism of action and effects of DES based primarily on different potentials as a chemical substance, pharmaceutical or hormonal agent. One well characterized model for DES exposure is the neonatal mouse model in which female and male pups are treated with DES (2µg/day) for the first five days of life and aged up to 18 months. DES treatment of female mice in this model produces a high incidence of reproductive tract tumors.11,12 The neonatal mouse model and other previously utilized rodent models were limited in that they could not elucidate the role of the estrogen receptor (ER) or DES as a hormonal substance in DES-induced toxicity.
The generation of mice lacking either estrogen receptor α (ERα) or estrogen receptor β (ERβ), i.e. ER knockout (ERKO) mice, and their incorporation into the neonatal mouse model of DES exposure allowed for examination of the role of the ER subtypes in mediating the effects of DES in vivo.13,14 Wild type (WT) female mice treated neonatally with DES displayed characteristic DES-induced reproductive tract lesions (Table 1 ).15 Treatment resulted in atrophy, smooth muscle disorganization, hyalinization, squamous metaplasia of the glandular epithelium, and endometrial hyperplasia. In the vagina, persistent epithelial cornification and vaginal adenosis were observed in a small percentage of animals following DES treatment. Finally, DES induced progressive proliferative lesions of the epithelium in the oviduct. None of these reproductive tract lesions were observed in identically treated αERKO female mice, indicating that DES toxicity is mediated via an ERα-dependent signaling pathway.15,16
Similar experiments utilizing the neonatal mouse model of DES exposure were conducted with WT, αERKO, and βERKO male mice to determine if DES-induced effects in the male reproductive tract are ER-mediated. When WT males are exposed neonatally to DES, they exhibit significant decreases in seminal vesicle size at all ages, while no changes in seminal vesicle size are observed in identically treated αERKO males and males treated with oil alone (Figure 1).17 With respect to the prostate gland which expresses clearly measureable amounts of both ER? and ER?, DES treatment of neonatal WT and ?ERKO mice produced histological changes in the aged ventral and dorsolateral prostates indicative of the phenotype of an estrogenized prostate gland, including increased stromal mass, epithelial hyperplasia and dysplasia, and interstitial lymphocyte infiltration.18 In addition, DES induced abnormal epithelial cell differentiation, indicative of estrogen imprinting in the dorsolateral prostate of both WT and βERKO mice. Observed abnormalities include the presence of a continuous layer of basal cells lining the epithelial ducts and acini, luminal cell hyperplasia, and loss of expression of dorsolateral prostate-2 protein, a functional differentiation marker in the dorsolateral prostate.17 None of these effects were observed in the prostate glands of identically treated αERKO mice, indicating that a functional ERα is required for DES-mediated activity. Collectively, these data suggest that DES elicits its toxic effects in the male reproductive tract through an ERα-mediated mechanism.
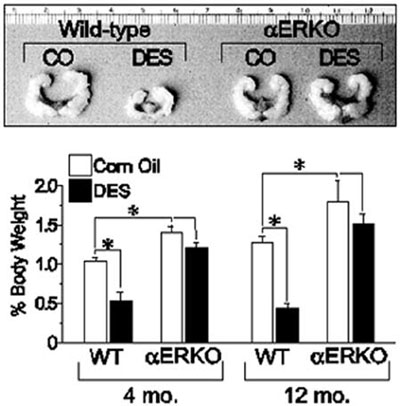
Figure 1. Effect of neonatal DES exposure on seminal vesicle weight in adult WT and αERKO mice. Top, seminal vesicles from 6-month old WT and αERKO mice following neonatal exposure to either vehicle (corn oil, CO) or DES (scale is in centimeters). Bottom, quantitative analysis of seminal vesicle weights from WT and αERKO mice at 4 and 12 months of age following neonatal exposure to CO or DES. *, P<0.05. (Modified from Prins, G.S., Birch, L.,Couse, J.F., Choi, I., Katzenellenbogen, B., and Korach, K.S., 2001 Cancer Res., 61: 6089-6097.)
Having confirmed the necessary role of ERα in mediating the DES-induced phenotypic changes in the reproductive tracts of female and male mice, studies have focused on elucidating the mechanisms underlying these changes. One area of focus in this regard has been the examination of DES-induced changes in the expression of genes in the Hox and Wnt families that are known to be critical for the normal differentiation and organization of the reproductive tract. Several lines of evidence suggest that altered gene expression in the Hox and Wnt families may be central to the DES-induced effects on the reproductive tract. First, their expression in the paramesonephric duct is essential for the development and organization of the female reproductive tract.19 Second, null mouse models of the Hox and Wnt gene families exhibit reproductive tract phenotypes similar to those of DES-treated mice.20-23 Finally, DES treatment of neonatal mice reduces the expression of Hoxa9, Hoxa10, Hoxa11, and Wnt7a in the female reproductive tract.22,24,25 Based on this evidence, studies were conducted with αERKO mice in the neonatal mouse model of DES exposure to determine the role of ERα in mediating DES-induced decreases in Hox and Wnt gene expression. Results from these studies showed that the uteri of 5-day old WT mice neonatally exposed to DES had an approximately 50% reduction in Hoxa10 and Hoxa11 mRNA levels, and a greater than 80% reduction in Wnt7a mRNA levels when compared to their expression levels in the uteri of control animals.15 In the uteri of DES treated αERKO mice, the expression levels of Hoxa10, Hoxa11, and Wnt7a were similar to those measured in the uteri of control mice, indicating that ERα mediates the DES-induced reduction in expression of these genes (Figure 2). These results substantiate the central role that ERα plays in mediating the in vivo effects of DES and provide further support for the argument that DES-induced alterations in Hox and Wnt gene expression are at least partially responsible for the reproductive tract abnormalities observed in DES-treated mice.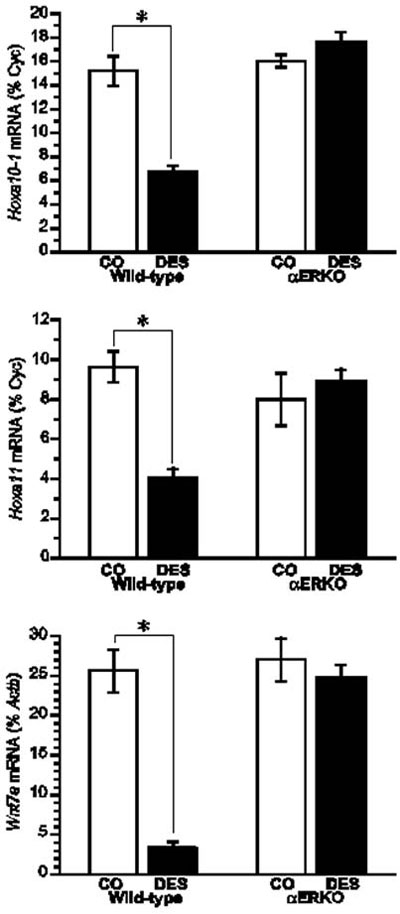
Figure 2. Effect of neonatal DES exposure on uterine expression of Hoxa10, Hoxa11, and Wnt7a in WT and αERKO mice. Top and middle, Quantitative analyses of ribonuclease protection assays (Hoxa10 and Hoxa11) (+/- SEM) showing average percentage of cyclophilin (normalization mRNA) for each treatment group. Bottom, Quantitative analysis of semiquantitative RT-PCR showing the average Wnt7a levels as a percentage of Actb (+/- SEM) for each treatment group. *, P<0.01. (Modified from Couse, J.F., Dixon, D., Yates, M., Moore, A.B., Ma, L., Maas, R., and Korach, K.S., 2001 Dev. Biol., 238: 224- 238.).
The molecular mechanisms underlying DES-induced decreases in Hox and Wnt gene expression have yet to be fully characterized. One proposed mechanism put forth involved epigenetic modifications such as DNA methylation, since DES increases lactoferrin and c-fos gene expression in the mouse uterus through demethylation of the lactoferrin promoter and hypomethylation of exon 4 of the c-fos gene.26,27 Subsequent studies showed that DES had no effect on promoter methylation of the Hoxa10 and Hoxa11 genes, indicating that DES utilizes a different mechanism to decrease expression of these genes.28 Subsequent studies examining the Hoxa10 promoter identified a nonconsensus estrogen response element (ERE) that is differentially regulated by estradiol and DES.29 Results from these studies showed that the maximal estradiol-induced luciferase activity was four times greater than that induced by DES in cells transfected with a luciferase reporter plasmid containing the Hoxa10 nonconsensus ERE. These results suggest that the nonconsensus Hoxa10 ERE is induced in a ligand-specific manner, which may be due to distinct conformational changes in the ER as a result of the unique ligand structures and/or the Hoxa10 nonconsensus ERE. Furthermore, these results may provide the mechanistic explanation for decreased Hoxa10 expression and the subsequent distinct phenotypes observed in the female reproductive tract following neonatal DES treatment. Whether a similar mechanism is responsible for DES-induced downregulation of Hoxa11 and Wnt7a expression remains to be determined.
GENISTEIN
Members of the phytoestrogen family of nonsteroidal, plant-derived compounds modulate ER signaling and therefore are potentially endocrine disrupting chemicals.30 The potential clinical application of phytoestrogens for prevention of hormone-dependent cancers and for lowering cholesterol levels has prompted extensive research aimed at elucidating the physiological effects of this class of compounds. The isoflavone genistein is a member of the phytoestrogen family and is found at high concentrations in soy-based products. Both in vitro and in vivo studies have clearly demonstrated that genistein exhibits estrogenic activity and tyrosine kinase inhibitory activity; however, the potential therapeutic benefits of genistein are not clear due to conflicting study results.31-34 For example, genistein has been shown to reduce mammary cancer rates in rats in some studies, while in other studies it has been shown to increase mammary cancer rates in rats.35-37 Additional animal studies have linked genistein to diminished reproductive capacity38,39 and, in a neonatal mouse model, genistein has been shown to induce uterine adenocarcinomas.40 The potential endocrine disrupting activity of genistein, coupled with the knowledge that humans are exposed to genistein through their diet, has stimulated interest in characterizing the biological effects of genistein on humans. While genistein has been shown to lower cholesterol levels in humans, little else is known regarding the beneficial and/or detrimental effects of human exposure to genistein.41,42 Exposure to genistein in infancy is of particular interest, as infants fed soy formula consume 6-9 mg/kg per day of isoflavones, approximately 65% of which is genistein, compared to 1 mg/kg per day for adults.43 This genistein level is 6-11 times higher than the amount reported to produce hormonal effects that alter the menstrual cycle of adult women.44 The presence of genistein in the human diet and the lack of knowledge regarding its definitive impact on human health have prompted investigations utilizing animal models to elucidate the mechanism of action of genistein and the biological consequences of exposure.
To elucidate the mechanistic aspects of genistein signaling, estrogen receptor binding assays have been performed to determine if genistein directly binds to ER. Results from these analyses show that genistein has a 20- to 30-fold higher affinity for ERβ than it does for ERα.45,46 However, in vitro assays demonstrate that genistein has only a slight preference for transactivation of gene expression through ERβ when compared to ERα, indicating that genistein is able to signal through both ERα and ERβ.46 To clarify the role of the ER subtypes in genistein signaling in vivo, neonatal mouse models have been utilized to examine its effects on the uterus, which predominantly expresses ERα. In this model, genistein elicited a classic uterotropic response, including increased uterine wet weight, epithelial cell height, gland number, and lactoferrin expression.47 Given the predominant expression of ERα in the mouse uterus, these results indicate that genistein can effectively signal through ERα in vivo. Further support for the role of ERα in genistein signaling is derived from studies showing that genistein activates the IGF-1 signaling pathway in the mouse uterus via an ERα-dependent mechanism.48 Collectively, these data demonstrate that ERα mediates genistein signaling in the mouse uterus. Subsequent studies in αERKO mice directly demonstrated that ERα is necessary for genistein-induced uterotropic responses. In these studies, estradiol and genistein significantly increased uterine wet weights in ovariectomized WT mice, while neither compound increased uterine wet weights in αERKO mice (Figure 3, unpublished data).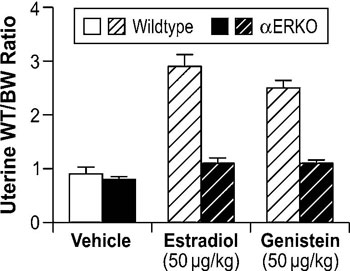
Figure 3. Role of ERα in modulating genistein-induced uterotropic effects. Ovariectomized WT and αERKO mice (n=4 mice/treatment group) were treated daily with subcutaneous injections of corn oil (vehicle), 10µg/kg estradiol, or 50µg/kg genistein for three days. On the fourth day whole body weights were measured and uteri were collected and weighed. The data represent uterine wet weight with respect to whole body weight. (Unpublished data from Lindzey, J. and Korach, K.S.).
The mouse ovary, in contrast to the uterus which predominantly expresses ERα, expresses both ERα and ERβ and provides a suitable model to determine the role of ERβ in mediating genistein signaling. As a result, in vivo studies have been performed to examine the effect of genistein on the mouse ovary. Results from these studies demonstrated that genistein induced a dose-dependent increase in multi-oocyte follicles in WT CD-1 mice treated for 5 days with genistein when compared to vehicle controls (Table 2 ).49 Given the established tyrosine kinase inhibitory activity of genistein, the nonestrogenic tyrosine kinase inhibitor lavendustin A was incorporated into these experiments to confirm that the results were due solely to the associated estrogenic activity of genistein. No multi-oocyte follicles were observed in mice treated with two different concentrations of lavendustin A, indicating that the genistein-induced increases in multi-oocyte follicles were due to its estrogenic activity and not to tyrosine kinase inhibition. Further experiments were conducted in C57BL/6, αERKO, and βERKO mice to determine if genistein signaling in the ovary is mediated by ERα, ERβ or a non-receptor-mediated mechanism. Results from these studies showed that genistein induced a concentration-dependent increase in multi-oocyte follicles in C57BL/6 and αERKO mice, while the incidence of multi-oocyte follicles in βERKO mice was significantly lower.49 These results indicate that ERβ is necessary for the induction of multi-oocyte follicles in genistein-treated neonatal mice.
Taken together, these in vivo data show that genistein utilizes both ERα- and ERβ-mediated mechanisms, depending on the specific tissue, to elicit estrogenic effects on the female mouse reproductive tract. While these models have proven useful in uncovering the effects and mechanism of action of genistein on the mouse reproductive tract, extrapolation of these results to humans has been hampered by conflicting data obtained from epidemiological studies. Therefore, continued efforts to elucidate the biological effects and mechanism of action of genistein are needed, as are further epidemiological studies, to obtain a clearer picture of the beneficial and detrimental effects of genistein exposure in humans.
METHOXYACETIC ACID
Methoxyacetic acid (MAA) is the major metabolite of the industrial solvent ethylene glycol monomethyl ether (EGME), used in paints, varnishes, dyes, and fuel additives. Occupational exposure to EGME, and consequently to its major metabolite MAA, have been associated with reproductive toxicity. In women, exposure to ethylene glycol ethers has been associated with increased risk of spontaneous abortion and subfertility, while it is associated with decreased sperm counts in exposed males.50-56 Laboratory studies have demonstrated that EGME targets the ovarian luteal cell, suppresses cyclicity, and inhibits ovulation in female rats, while in male rats it reduces testicular size and fertility.57,58 Subsequent studies have established that MAA is responsible for the reproductive toxicities associated with EGME exposure.53,57
MAA is a member of the short-chain fatty acid family, which includes the antiepileptic drug valproic acid (VPA) and the intestinal bacterial product sodium butyrate (NaB). Interestingly, VPA has also been associated with reproductive toxicities, including menstrual abnormalities and polycystic ovaries.59-61 One shared feature of MAA, VPA, and NaB is their ability to inhibit histone deacetylases and alter gene expression via histone hyperacetylation.62-66 However, what role this effect plays in the reproductive toxicities associated with MAA and VPA is unknown.
The reproductive toxicities associated with MAA exposure in humans and animals coupled with the critical role of estrogen signaling in normal male and female reproductive function suggest that MAA may disrupt normal estrogen signaling. In addition, the aforementioned reproductive toxicities are similar to the reproductive phenotypes observed in αERKO and aromatase knockout mice, further suggesting that MAA may disrupt estrogen signaling. Therefore, studies have been conducted to characterize the effects of MAA on estrogen signaling in vitro and in vivo. MAA has been shown to enhance exogenous ER signaling in vitro by transiently transfecting an ER expression vector and a luciferase reporter into HeLa cells and treating them with estradiol in the presence and absence of MAA.62 These results have been confirmed in our laboratory by transfecting ER-negative HeLa cells with an expression vector for either ERα or ERβ and the estrogen-inducible 3x-ERE-TATA-Luc firefly luciferase reporter plasmid. As shown in Figure 4A and 4B, MAA potentiates the activity of estradiol in HeLa cells transfected with the luciferase reporter and either ERα or ERβ.67 These data are incongruent with the reproductive toxicities observed following MAA exposure; therefore, additional experiments were performed to examine the effect of MAA on endogenous ER signaling. Interestingly, when ER-positive MCF-7 cells were transfected with the 3x-ERE-TATA-Luc reporter plasmid, but no ER expression vector, and treated identically to the HeLa cells, no potentiation of E2-induced luciferase activity was observed (data not shown).67 However, when an ER expression vector was cotransfected into the MCF-7 cells, MAA was able to potentiate the estrogen-stimulated luciferase activity. These results demonstrate that MAA differentially affects exogenous and endogenous ER signaling. Further experiments showed that MAA dose-dependently increases exogenous ERα protein expression levels in HeLa cells in the presence or absence of estradiol, which is due to activation of the CMV promoter contained in the expression vectors for both ERα and ERβ (Figure 4C, 4D).67 In contrast to its effects on the exogenous ER, MAA decreases endogenous ERα protein and mRNA expression levels by approximately 50% in MCF-7 cells treated with 5mM MAA for 24h when compared to vehicle controls (Figure 5A and 5B).67 A similar effect was observed in the uteri of ovariectomized C57BL/6 mice treated with 400mg/kg MAA for 2.5h, as ERα mRNA levels decreased by approximately 30% following MAA treatment when compared to vehicle controls.67 Taken together, these data illustrate that MAA has similar effects on endogenous ERα expression in vitro and in vivo.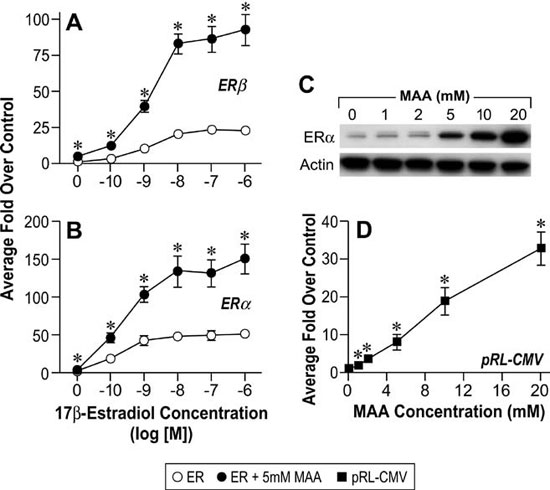
Figure 4. MAA activates the Cytomegalovirus CMV) promoter in vitro. A. HeLa cells were transiently transfected with a human ERβ expression vector, the estrogen-responsive 3X-ERE-TATA-Luc reporter plasmid, and the CMV-β-gal reporter plasmid and treated for 18h with either vehicle, increasing concentrations of E2, 5mM MAA, or increasing concentrations of E2 plus 5mM MAA. The histogram represents the average fold over control of duplicate samples from three independent experiments. The error bars represent the standard error of the mean (SEM), *p<0.05 comparing identical treatments +/- 5mM MAA. B. HeLa cells were transiently transfected with a human ERα expression vector, 3X-ERE-TATALuc and CMV-β-gal and treated identically to the cells in Figure 1A. The data shown represent the average fold over control of duplicate samples from three independent experiments and the error bars indicated the SEM, *p<0.05 comparing identical treatments +/- 5mM MAA. C. HeLa cells were transiently transfected with a human ERα expression vector and treated with either vehicle or increasing concentrations of MAA for 18h. Total protein was isolated from the cells and ERα protein expression was analyzed by Western blot. The data shown are representative of three independent experiments. D. HeLa cells were transiently transfected with the pRL-CMV reporter plasmid and treated with either vehicle or increasing concentrations of MAA for 18h. The data shown represent the average fold over control of duplicate samples from three independent experiments. The error bars represent the SEM, *p<0.01 comparing treatment with MAA to vehicle control. (Modified from Henley, D.V., Mueller, S., and Korach, K.S. In Press Environ Health Persp.).
Subsequent to these studies, gene expression experiments were performed to determine if the observed decrease in ERα expression correlated with alterations in estrogen signaling. In MCF-7 cells, pre-treatment with 5mM MAA followed by 1nM estradiol attenuated the expression of the estrogen-regulated genes pS2, MYC, SPUVE, GREB1, and MCM3 when compared to treatment with 1nM estradiol alone (Figure 6).67 MAA had a similar effect on estrogen-modulated gene expression in vivo, as ovariectomized C57BL/6 mice treated with 400mg/kg MAA and 1µg/kg estradiol had decreased uterine expression of the estrogen-inducible genes Greb1, Inhbb, and Fos when compared to uterine expression in mice treated with estradiol alone.67 These studies show that MAA attenuates estradiol-mediated signaling in vitro and in vivo and illustrate the functional consequences of MAA-induced decreases in ERα expression. The MAA-mediated attenuation of estrogen-inducible gene expression is likely due to the observed decrease of ER expression following MAA treatment, as binding experiments have demonstrated that MAA does not compete with estradiol for binding to the ER.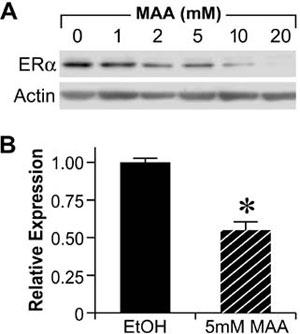
Figure 5. Methoxyacetic Acid (MAA) decreases endogenous ERα expression. A. MCF-7 cells were treated for 24h with either vehicle (EtOH, Ethanol) or increasing concentrations of MAA. Total protein was isolated from each sample and analyzed for ERα protein expression by Western blot. The data shown are representative of results from three independent experiments. B. MCF-7 cells were treated for 24h with either vehicle or 5mM MAA. Total RNA was isolated from each sample and steady state mRNA levels of ERα were measured by real-time PCR. The data shown represent the average fold over control obtained from duplicate samples in four independent experiments, and the error bars indicate the SEM, *p<0.01 comparing treatment with 5mM MAA to vehicle control. (Modified from Henley, D.V., Mueller, S., and Korach, K.S. In Press Environ Health Persp.).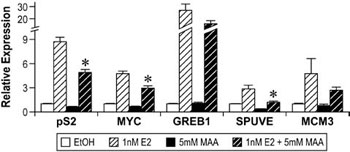
Figure 6. Methoxyacetic Acid (MAA) disrupts estrogen-mediated endogenous gene expression. MCF-7 cells were pretreated with either vehicle (EtOH, Ethanol) or 5mM MAA for 2h and then treated for 18h with either vehicle or 1nM E2. Total RNA was isolated from the samples and the expression of endogenous estrogen-responsive genes was analyzed by real-time PCR. The data shown represent the average fold over control of duplicate samples from at least three independent experiments. Error bars represent the SEM, *p<0.01 comparing treatment with 1nM E2 + 5mM MAA to treatment with 1nM E2 alone. The error bars represent SEM. (Modified from Henley, D.V., Mueller, S., and Korach, K.S. In Press Environ Health Persp.).
Collectively, these data confirm that MAA disrupts ER signaling through a mechanism that involves decreased endogenous ERα expression. These results are consistent with the reproductive toxicities associated with EGME exposure and provide some insight into the mechanism of action of MAA with regard to reproductive toxicity, which, based on these results, include disrupted ER signaling as a result of decreased ER expression. These results also provide a framework for future studies to examine the impact of MAA on ER expression and signaling in other parts of the reproductive tract and to further pursue the mechanism(s) of action underlying MAA-induced reproductive toxicity.
LAVENDER OIL AND TEA TREE OIL
Lavender oil and tea tree oil are essential oils frequently used in over-the-counter health and beauty products, including topically applied lotions, gels, and creams, to impart pleasant aromas and for a variety of purported health benefits. Consequently, topical exposure to essential oils is widespread; however, little is known regarding the acute and long-term effects of such exposure in humans. Clinical evidence suggesting that lavender oil and tea tree oil may possess endocrine disrupting activity first surfaced with the report of three patients presenting with idiopathic male prepubertal gynecomastia, all of whom had topically applied products containing either lavender oil or a combination of lavender and tea tree oil.68 Gynecomastia is clinically characterized by enlarged breast tissue and is the result of disrupted estrogen and androgen signaling which lead to increased or unopposed estrogen action on breast tissue.69 Although pubertal male gynecomastia is common, an underlying cause is not identified in most cases (idiopathic). Hence pubertal gynecomastial does not usually require investigation. Pre-pubertal gynecomastia, nevertheless, is rare and must be investigated. This is the reason why the three boys were thoroughly investigated for the gynecomastia.70,71 These three male prepubertal patients with gynecomastia had normal low serum levels of endogenous sex steroids and no apparent underlying pathologies. One common feature among the three boys was that each of them had topically applied over-the-counter products containing lavender oil, and in one case a product contained both lavender oil and tea tree oil. In all three boys, the onset of gynecomastia coincided with the topical application of these products and resolved with the discontinuation of their use, suggesting that lavender oil, and perhaps tea tree oil, may possess endocrine disrupting activity that leads to the development of gynecomastia.
These clinical case reports, coupled with the link between gynecomastia and disrupted sex steroid signaling, prompted studies to characterize the effects of lavender oil and tea tree oil on estrogen and androgen signaling. Estrogenic activity was assessed by conducting dose-response experiments in MCF-7 cells transfected with the estrogen-inducible 3x-ERE-TATA-Luc reporter plasmid. Both lavender oil and tea tree oil induced ERE-dependent luciferase activity in a dose-dependent manner, with maximal activity observed at 0.025% vol/vol for each oil that corresponded to approximately 50% of the activity induced by 1nM estradiol (Figure 7).68 This effect was attenuated in the presence of the pure ER antagonist fulvestrant, indicating that the estrogenic activity of the oils is ER-dependent. Further studies showed that both lavender oil and tea tree oil modulate the endogenous expression of the estrogen-responsive genes MYC, CTSD, and IGFBP-3 in MCF-7 cells in a manner similar to that observed with 1nM estradiol with respect to both the magnitude and timing of the response (Figure 8).68 These responses were ER-dependent as well, as they were attenuated in the presence of fulvestrant. The estrogenic activity of tea tree oil has been independently confirmed in a separate study that examined the effect of tea tree oil in MCF-7 proliferation assays.72 Results from these studies show that tea tree oil induced a significant dose-dependent estrogenic response in vitro, with the maximal proliferative response occurring at a concentration of 0.0125% that corresponded to 34% of the maximal estradiol response in the assays.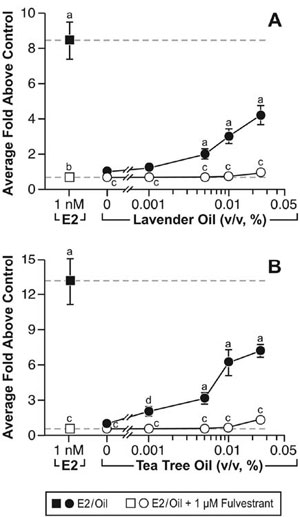
Figure 7. In vitro estrogenic activity of lavender oil and tea tree oil in reporter gene assays. MCF-7 cells were transiently transfected with the estrogen-inducible 3x-ERE-luciferase reporter plasmid and treated with increasing concentrations of either lavender oil or tea tree oil for 18 hours in the presence and absence of fulvestrant, an ER antagonist. Estradiol treatment (1 nM) was included as a positive control. Data are plotted as fold-luciferase activity over vehicle control. (Modified from Henley, D.V., Lipson, N., Korach, K.S., and Bloch, C.A., NEJM, 356, 481, 2007).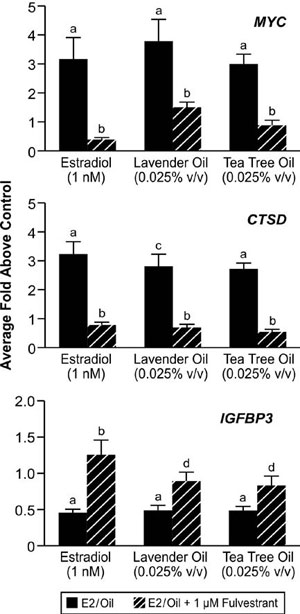
Figure 8. In vitro effects of lavender oil and tea tree oil on the expression of estrogen-regulated endogenous genes. MCF-7 cells were treated with 1 nM estradiol, 0.025% lavender oil, or 0.025% tea tree oil in the presence or absence of fulvestrant and real-time PCR was performed to measure changes in the expression of the estrogen-inducible genes MYC, IGFBP3, and CTSD. Data shown are normalized and relative to a vehicle control. (Modified from Henley, D.V., Lipson, N., Korach, K.S., and Bloch, C.A., NEJM, 356, 481, 2007).
Since altered androgen signaling can also contribute to the development of gynecomastia, studies were conducted to characterize the effect of lavender oil and tea tree oil on androgen signaling. The MDA-kb2 breast cancer cell line, which expresses AR and has been stably transfected with an androgen-inducible mouse mammary tumor virus promoter, was incorporated into these experiments to evaluate the androgenic activity of both oils.73 While increased luciferase activity was observed following treatment of MDA-kb2 cells with the AR agonist dihydrotestosterone (DHT), no increase in luciferase activity was observed following exposure to lavender oil or tea tree oil regardless of concentration, indicating that the oils do not possess androgenic activity.68 However, both oils elicited a concentration-dependent inhibition of DHT-induced luciferase activity, indicating that both oils possess antiandrogenic activity (Figure 9).68 This antiandrogenic effect extended to the DHT-induced expression of the androgen-responsive endogenous genes CYP4F8, C1orf116, UGT2B28, and SEC14L2 in MDA-kb2 cells treated with 0.1nM DHT and either lavender oil or tea tree oil (Figure 10).68 Expression levels of AR mRNA and protein were not altered by either oil, indicating that the observed antiandrogenic effect of the oils is not due to downregulation of AR expression.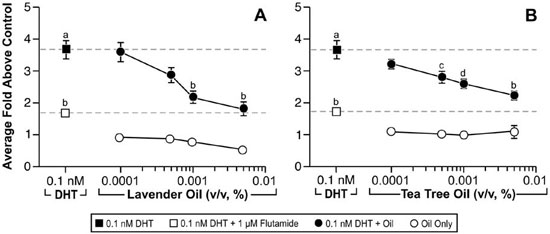
Figure 9. In vitro antiandrogenic activity of lavender oil and tea tree oil in reporter gene assays. MDA-kb2 cells stably transfected with the MMTV-luciferase reporter plasmid were treated for 24 hours with increasing concentrations of lavender oil or tea tree oil in the presence or absence of 0.1 nM dihydrotestosterone (DHT). The androgen receptor antagonist flutamide (1 µM) was included as a control antagonist for the androgen receptor. Luciferase activity was measured and data plotted as the average fold above control. (Modified from Henley, D.V., Lipson, N., Korach, K.S., and Bloch, C.A., NEJM, 356, 483, 2007).
Taken together, these data illustrate that both lavender and tea tree oil possess estrogenic and anti-androgenic properties, and this activity may underlie the onset of prepubertal gynecomastia in the three patients described earlier with idiopathic prepubertal gynecomastia. The endocrine disrupting activity of these oils may be shared with other essential oils as well, as estrogenic and antiandrogenic properties have been reported for other essential oils and some of their monoterpene constituents.74-79 In light of these data and the widespread use of essential oils in over-the-counter health and beauty products, additional studies should be conducted to more fully characterize their endocrine disrupting properties. One prominent challenge with respect to such studies is that essential oils contain numerous components, most of which are monoterpenes. Therefore, identifying which component or components of the oils possess the endocrine disrupting activity is difficult. However, identification of these components would greatly facilitate identification of other oils that may possess similar endocrine disrupting activity, as their presence in an essential oil would suggest potential endocrine disrupting activity.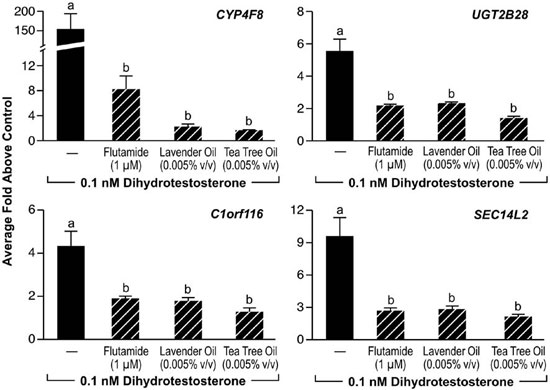
Figure 10. In vitro effects of lavender oil and tea tree oil on androgen-stimulated endogenous gene expression. MDA-kb2 cells were treated with 0.1 µM dihydrotestosterone for 24 hours in the presence or absence of 0.0005% lavender oil, 0.0005% tea tree oil, or 1 ?M flutamide and real-time PCR was performed to measure changes in the expression of androgen-inducible genes. Data shown are normalized and relative to a vehicle control. (Modified from Henley, D.V., Lipson, N., Korach, K.S., and Bloch, C.A., NEJM, 356, 483, 2007).
CONCLUSIONS
In this review we have provided examples of the activity and actions of several different EDC type compounds of varying chemical structure, properties, and sources. Findings from in vitro cell based assays and/or experimental animal studies have provided evidence of their hormonal biological actions involving receptor mediated activities as a mechanism for their toxicities. Additionally, an example is given of a unique EDC clinical observation regarding the properties of some essential oils. As expected, further observations of other types of toxicities and development of additional experimental models and systems will be required for the evaluation of the mechanisms involved. Finally, the impact that these exposures may have on the human population will require appreciation of potential chemical effects from certain exposure as well as broader epidemiological assessments in support of the experimental findings.
REFERENCES
1. Dodds EC, Lawsaon W, Robinson R, 1938 Estrogenic activity of certain synthetic compounds. Nature 141247-141248.
2. Smith OW, Smith GBS, Hurwitz D, 1946 Increased excretion of pregnanediol in pregnancy from diethylstilbestrol with special reference to the prevention of late pregnancy accidents. Med Rec Ann 40: 1669-1671.
3. Dieckmann WJ, Davis ME, Rynkiewicz LM, Pottinger RE, Gabbe SG, 1999 Does the administration of diethylstilbestrol during pregnancy have therapeutic value 1953? Am J Obstet Gynecol 66:1062-1081.
4. Herbst AL, Ulfelder H, Poskanzer DC, 1971 Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med 284: 878-881.
5. Melnick S, Cole P, Anderson D, Herbst A, 1987 Rates and risks of diethylstilbestrol-related clear-cell adenocarcinoma of the vagina and cervix. An update. N Engl J Med 316: 514-516.
6. Herbst AL, 2000 Behavior of estrogen-associated female genital tract cancer and its relation to neoplasia following intrauterine exposure to diethylstilbestrol (DES). Gynecol Oncol 76: 147-156.
7. Mittendorf R, 1995 Teratogen update: carcinogenesis and teratogenesis associated with exposure to diethylstilbestrol (DES) in utero. Teratology 51: 435-445.
8. Kaufman RH, Adam E, Hatch EE, et al, 2000 Continued follow-up of pregnancy outcomes in diethylstilbestrol-exposed offspring. Obstet Gynecol 96: 483-489.
9. Giusti RM, Iwamoto K, Hatch EE, 1995 Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med 122: 778-788.
10. Marselos M, Tomatis L, 1992 Diethylstilboestrol: I, Pharmacology, Toxicology and carcinogenicity in humans. Eur J Cancer 28A: 1182-1189.
11. Newbold RR, McLachlan JA, 1982 Vaginal adenosis and adenocarcinoma in mice exposed prenatally or neonatally to diethylstilbestrol. Cancer Res 42: 2003-2011.
12. Marselos M, Tomatis L, 1992 Diethylstilboestrol: II, pharmacology, toxicology and carcinogenicity in experimental animals. Eur J Cancer 29A: 149-55.
13. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O, 1993 Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 90: 11162-11166.
14. Krege JH, Hodgin JB, Couse JF, et al, 1998 Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci USA 95: 15677-15682.
15. Couse JF, Dixon D, Yates M, et al, 2001 Estrogen receptor-alpha knockout mice exhibit resistance to the developmental effects of neonatal diethylstilbestrol exposure on the female reproductive tract. Dev Biol 238: 224-238.
16. Couse JF, Korach KS, 2004 Estrogen receptor-alpha mediates the detrimental effects of neonatal diethylstilbestrol (DES) exposure in the murine reproductive tract. Toxicology 205: 55-63.
17. Prins GS, Birch L, Couse JF, Choi I, Katzenellenbogen B, Korach KS, 2001 Estrogen imprinting of the developing prostate gland is mediated through stromal estrogen receptor alpha: studies with alphaERKO and betaERKO mice. Cancer Res 61: 6089-6097.
18. Singh J, Handelsman DJ, 1999 Morphometric studies of neonatal estrogen imprinting in the mature mouse prostate. J Endocrinol 162: 39-48.
19. Doll? P, Izpis?a-Belmonte JC, Brown JM, Tickle C, Duboule D, 1991 HOX-4 genes and the morphogenesis of mammalian genitalia. Genes Dev 5: 1767-1767.
20. Hsieh-Li HM, Witte DP, Weinstein M, et al, 1995 Hoxa 11 structure, extensive antisense transcription, and function in male and female fertility. Development 121: 1373-1385.
21. Satokata I, Benson G, Maas R, 1995 Sexually dimorphic sterility phenotypes in Hoxa10-deficient mice. Nature 374: 460-463.
22. Miller C, Degenhardt K, Sassoon DA, 1998 Fetal exposure to DES results in de-regulation of Wnt7a during uterine morphogenesis. Nat Genet 20: 228-230.
23. Parr BA, McMahon AP, 1998 Sexually dimorphic development of the mammalian reproductive tract requires Wnt-7a. Nature 395: 707-710.
24. Block K, Kardana A, Igarashi P, Taylor HS, 2000 In utero diethylstilbestrol (DES) exposure alters Hox gene expression in the developing mullerian system. Faseb J 14: 1101-1108.
25. Ma L, Benson GV, Lim H, Dey SK, Maas RL, 1998 Abdominal B (AbdB) Hoxa genes: regulation in adult uterus by estrogen and progesterone and repression in mullerian duct by the synthetic estrogen diethylstilbestrol (DES). Dev Biol 197: 141-154.
26. Li S, Washburn KA, Moore R, et al, 1997 Developmental exposure to diethylstilbestrol elicits demethylation of estrogen-responsive lactoferrin gene in mouse uterus. Cancer Res 57: 4356-4359.
27. Li S, Hansman R, Newbold R, Davis B, McLachlan JA, Barrett JC, 2003 Neonatal diethylstilbestrol exposure induces persistent elevation of c-fos expression and hypomethylation in its exon-4 in mouse uterus. Mol Carcinog 38: 78-84.
28. Li S, Ma L, Chiang T, et al, 2001 Promoter CpG methylation of Hox-a10 and Hox-a11 in mouse uterus not altered upon neonatal diethylstilbestrol exposure. Mol Carcinog 32: 213-219.
29. Akbas GE, Song J, Taylor HS, 2004 A HOXA10 estrogen response element (ERE) is differentially regulated by 17 beta-estradiol and diethylstilbestrol (DES). J Mol Biol 340: 1013-1023.
30. Moutsatsou P, 2007 The spectrum of phytoestrogens in nature: our knowledge is expanding. Hormones (Athens) 6: 173-193.
31. Santell RC, Chang YC, Nair MG, Helferich WG, 1997 Dietary genistein exerts estrogenic effects upon the uterus, mammary gland and the hypothalamic/pituitary axis in rats. J Nutr 127: 263-269.
32. Hsieh CY, Santell RC, Haslam SZ, Helferich WG, 1998 Estrogenic effects of genistein on the growth of estrogen receptor-positive human breast cancer (MCF-7) cells in vitro and in vivo. Cancer Res 58: 3833-3838.
33. Collins BM, McLachlan JA, Arnold SF, 1997 The estrogenic and antiestrogenic activities of phytochemicals with the human estrogen receptor expressed in yeast. Steroids 62: 365-372.
34. Akiyama T, Ishida J, Nakagawa S, et al, 1987 Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem 262: 5592-5595.
35. Hilakivi-Clarke L, Onojafe I, Raygada M, et al, 1999 Prepubertal exposure to zearalenone or genistein reduces mammary tumorigenesis. Br J Cancer 80: 1682-1688.
36. Lamartiniere CA, Zhang JX, Cotroneo MS, 1998 Genistein studies in rats: potential for breast cancer prevention and reproductive and developmental toxicity. Am J Clin Nutr 68(6 Suppl): 1400S-1405S.
37. Hilakivi-Clarke L, Cho E, Onojafe I, Raygada M, Clarke R, 1999 Maternal exposure to genistein during pregnancy increases carcinogen-induced mammary tumorigenesis in female rat offspring. Oncol Rep 6: 1089-1095.
38. Bradbury RB, White DE, 1954 Estrogens and related substances in plants. Vitam Horm 12207-12233.
39. Setchell KD, Gosselin SJ, Welsh MB, et al, 1987 Dietary estrogens--a probable cause of infertility and liver disease in captive cheetahs. Gastroenterology 93: 225-233.
40. Newbold RR, Banks EP, Bullock B, Jefferson WN, 2001 Uterine adenocarcinoma in mice treated neonatally with genistein. Cancer Res 61: 4325-4328.
41. Potter SM, Baum JA, Teng H, Stillman RJ, Shay NF, Erdman JW Jr., 1998 Soy protein and isoflavones: their effects on blood lipids and bone density in postmenopausal women. Am J Clin Nutr 68: Suppl 6: 1375-1379.
42. Cruz ML, Wong WW, Mimouni F, et al, 1994 Effects of infant nutrition on cholesterol synthesis rates. Pediatr Res 35: 135-140.
43. Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE, 1997 Exposure of infants to phyto-oestrogens from soy-based infant formula. Lancet 350: 23-27.
44. Cassidy A, Bingham S, Setchell KD, 1994 Biological effects of a diet of soy protein rich in isoflavones on the menstrual cycle of premenopausal women. Am J Clin Nutr 60: 333-340.
45. Kuiper GG, Lemmen JG, Carlsson B, et al, 1998 Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139: 4252-4263.
46. Mueller SO, Simon S, Chaek, Metzler M, Korach CS, 2004 Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor alpha (ERalpha) and ERbeta in human cells. Toxicol Sci 80: 14-25.
47. Jefferson WN, Padilla-Banks E, Clark G, Newbold RR, 2002 Assessing estrogenic activity of phytochemicals using transcriptional activation and immature mouse uterotrophic responses. J Chromatogr B Analyt Technol Biomed Life Sci 777: 179-189.
48. Klotz DM, Hewitt SC, Korach KS, Diaugustine RP, 2000 Activation of a uterine insulin-like growth factor I signaling pathway by clinical and environmental estrogens: requirement of estrogen receptor-alpha. Endocrinology 141: 3430-3439.
49. Jefferson WN, Couse JF, Padilla-Banks E, Korach KS, Newbold RR, 2002 Neonatal exposure to genistein induces estrogen receptor (ER)alpha expression and multioocyte follicles in the maturing mouse ovary: evidence for ERbeta-mediated and nonestrogenic actions. Biol Reprod 67: 1285-1296.
50. Johanson G, 2000 Toxicity review of ethylene glycol monomethyl ether and its acetate ester. Crit Rev Toxicol 30: 307-345.
51. Chapin RE, Dutton SL, Ross MD, Lamb JC 4th, 1985 Effects of ethylene glycol monomethyl ether (EGME) on mating performance and epididymal sperm parameters in F344 rats. Fundam Appl Toxicol 5: 182-189.
52. Brinkworth MH, Weinbauer GF, Schlatt S, Nieschlag E, 1995 Identification of male germ cells undergoing apoptosis in adult rats. J Reprod Fertil 105: 25-33.
53. Foster PM, Creasy DM, Foster JR, Gray TJ, 1984 Testicular toxicity produced by ethylene glycol monomethyl and monoethyl ethers in the rat. Environ Health Perspect 57207-57217.
54. Nagano K, Nakayama E, Oobayashi H, et al, 1981 Embryotoxic effects of ethylene glycol monomethyl ether in mice. Toxicology 20: 335-343.
55. Correa A, Gray RH, Cohen R, et al, 1996 Ethylene glycol ethers and risks of spontaneous abortion and subfertility. Am J Epidemiol 143: 707-717.
56. El-Zein RA, Abdel-Rahman SZ, Morris DL, Legator MS, 2002 Exposure to ethylene glycol monomethyl either: clinical and cytogenetic findings. Arch Environ Health 57: 371-376.
57. Davis BJ, Almekinder JL, Flagler N, Travlos G, Wilson R, Maronpot RR, 1997 Ovarian luteal cell toxicity of ethylene glycol monomethyl ether and methoxy acetic acid in vivo and in vitro. Toxicol Appl Pharmacol 142: 328-337.
58. Rao PN, 1971 Preparation of 9-alpha,11-xi-tritiated 17-alpha-ethynylestradiol, mestranol, estradiol-alpha-17-beta, and norethindrone. Steroids 18: 219-229.
59. Isojarvi JI, Laatikainen TJ, Pakarinen AJ, Juntunen KT, Myllyl? W, 1993 Polycystic ovaries and hyperandrogenism in women taking valproate for epilepsy. N Engl J Med 329: 1383-1388.
60. O’Donovan C, Kusumakar V, Graves GR, Bird DC, 2002 Menstrual abnormalities and polycystic ovary syndrome in women taking valproate for bipolar mood disorder. J Clin Psychiatry 63: 322-330.
61. L?fgren E, Mikkonen K, Tolonen U, et al, 2007 Reproductive endocrine function in women with epilepsy: the role of epilepsy type and medication. Epilepsy Behav 10: 77-83.
62. Jansen MS, Nagel SC, Miranda PJ, Lobenhofer EK, Afshari CA, McDonnell DP, 2004 Short-chain fatty acids enhance nuclear receptor activity through mitogen-activated protein kinase activation and histone deacetylase inhibition. Proc Natl Acad Sci U S A 101: 7199-7204.
63. G?ttlicher M, Minucci S, Zhu P, et al, 2001 Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo J 20: 6969-6978.
64. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS, 2001 Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276: 36734-36741.
65. Sealy L, Chalkley R, 1978 The effect of sodium butyrate on histone modification. Cell 14: 115-121.
66. Boffa LC, Vidali G, Mann RS, Allfrey VG, 1978 Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem 253: 3364-3366.
67. Henley DV, Mueller S, Korach KS, 2009 The short-chain fatty acid methoxyacetic acid dirsupts endogenous estrogen receptor alpha mediated signaling. Environ Health Perspect.
68. Henley DV, Lipson N, Korach KS, Bloch CA, 2007 Prepubertal gynecomastia linked to lavender and tea tree oils. N Engl J Med 356: 479-485.
69. Braunstein GD, 1993 Gynecomastia. N Engl J Med 328: 490-495.
70. Einav-Bachar R, Phillip M, Aurbach-Klipper Y, Lazar L, 2004 Prepubertal gynaecomastia: aetiology, course and outcome. Clin Endocrinol (Oxf) 61: 55-60.
71. Descamps H, Chaussain JL, Job JC, 1985 [Gynecomastia in boys before puberty]. Arch Fr Pediatr 42: 87-89.
72. Nielsen JB, 2008 What you see may not always be what you get--bioavailability and extrapolation from in vitro tests. Toxicol In Vitro 22: 1038-1042.
73. Wilson VS, Bobseine K, Lambright CR, Gray LE Jr., 2002 A novel cell line, MDA-kb2, that stably expresses an androgen- and glucocorticoid-responsive reporter for the detection of hormone receptor agonists and antagonists. Toxicol Sci 66: 69-81.
74. Perry NS, Houghton PJ, Sampson J, et al, 2001 In-vitro activity of S. lavandulaefolia (Spanish sage) relevant to treatment of Alzheimer’s disease. J Pharm Pharmacol 53: 1347-1356.
75. Tabanca N, Khan SI, Bedir E, et al, 2004 Estrogenic activity of isolated compounds and essential oils of Pimpinella species from Turkey, evaluated using a recombinant yeast screen. Planta Med 70: 728-735.
76. Howes MJ, Houghton PJ, Barlow DJ, Pocock VJ, Milligan SR, 2002 Assessment of estrogenic activity in some common essential oil constituents. J Pharm Pharmacol 54: 1521-1528.
77. Dhar SK, 1995 Anti-fertility activity and hormonal profile of trans-anethole in rats. Indian J Physiol Pharmacol 39: 63-67.
78. Geldof AA, Engel C, Rao BR, 1992 Estrogenic action of commonly used fragrant agent citral induces prostatic hyperplasia. Urol Res 20: 139-144.
79. Chung BH, Lee HY, Lee JS, Young CY, 2006 Perillyl alcohol inhibits the expression and function of the androgen receptor in human prostate cancer cells. Cancer Lett 236: 222-228.
Address for correspondence:
Kenneth S. Korach, Ph.D., Receptor Biology Section, Laboratory of Reproductive and Developmental Toxicology,
National Institute of Environmental Health Sciences, National Institutes of Health, MD B3-02, P.O. Box 12233, Research
Triangle Park, NC 27709 USA,
Tel.: (919) 541-3512, Fax: (919) 541-0696, e-mail: korach@niehs.nih.gov
Received 10-02-10, Revised 20-05-10, Accepted 10-06-10