1Endocrine Unit, Second Department of Obstetrics and Gynecology, Aretaieion Hospital, 2Endocrine Section, First Department of Medicine, Laiko Hospital, 3First Department of Pediatrics, "Agia Sophia" Children's Hospital, Athens University Medical School, Athens, 115 27, Greece
Exercise represents a physical stress that challenges homeostasis. In response to this stressor, autonomic nervous system and the hypothalamic-pituitary-adrenal axis are known to react and to participate in the maintenance of homeostasis. This includes elevation of cortisol and cathecholamines in plasma. However, sustained physical conditioning in highly trained athletes is associated with a decreased hypothalamic-pituitary-adrenal response to exercise. On the other hand, highly trained athletes exhibit a chronic mild hypercortisolism at baseline that may be an adaptive change to chronic exercise. In addition the proinflammatory cytokine IL-6 is also activated. Moreover, exercise stimulates the secretion of GH and prolactin, and may influence the type of immunity by stimulating TH2 response profile. Besides, the stress of exercise inhibits the gonadal function, through the production of glucocorticoids and cathecholamines, as well as through activation of the CRH neurons. Nowadays, apart from the beneficial effects of exercise, there is increasing incidence of exercise-related short- and long- term consequences, especially concerning the female athlete that many authors describe as the so-called "exercise-related female reproductive dysfunction". These consequences include amenorrhea, infertility, eating disorders, osteoporosis, coronary heart disease and euthyroid "sick" syndrome. The mechanisms involved in the pathogenesis of the above disorders are discussed in this review.
Catecholamines, CRH, Cortisol, Exercise, GnRH, Proinflammatory cytokines, Vasopressin
1. INTRODUCTION
Exercise is nowadays playing an increasingly important role in the lifestyle of the Western world. Its undoubted beneficial effect on the health of the body, and consequently of the mind, has been underlined in numerous scientific studies. However, exercise in excess can be deleterious to certain organs and systems of the body. In fact, exercise is a stress situation for which the body must find a new dynamic equilibrium. This dynamic process requires, among other things, adaptive responses of the hormonal systems, which are examined in this review.
2. THE STRESS SYSTEM
The term "stress" describes the state of the organism under the influence of external or internal forces, or "stressors", which threaten to alter its dynamic equilibrium or homeostasis. The adaptive changes occurring in response to stressors are both behavioral and physical. Once a certain threshold has been exceeded, a systemic reaction takes place that involves the "stress system" in the brain along with its peripheral components, the hypothalamic-pituitary-adrenal (HPA) axis, and the autonomic sympathetic system1 (Figure 1).

Figure 1. Heuristic representation of the hypothalamic-pituitary-adrenal axis and the locus ceruleus/norepinephrine (LC/NE) sympathetic system interplay. The dotted lines represent inhibition while the solid lines represent stimulation.
The central players among these components of this system are the corticotropin-releasing hormone (CRH) and vasopressin (AVP) neurons in the paraventricular nucleus of the hypothalamus (PVN), and other brain areas, like the locus ceruleus (LC)/norepinephrine (NE) and central autonomic sympathetic system in the brainstem.
2.1. The HPA axis and the sympathetic system (Figure 1)
The principal central nervous system regulators of the HPA axis are CRH and AVP. Corticotropin-releasing hormone, the main ACTH secretagogue, is synthesized by parvicellular and magnocellular PVN neurons and is secreted along with AVP into the hypophyseal portal blood via their projecting axons to the median eminence and to the posterior pituitary, respectively. Corticotropin-releasing hormone is also synthesized by anterior pituitary corticotroph cells (Figure 1). Corticotropin-releasing hormone neurons also possess axons that terminate in the LC/NE-sympathetic system neurons in the brainstem. Neurons of the latter systems send projections, mostly noradrenergic, to the PVN. Thus, there are reciprocal interactions between the CRH neurons and those of the LC/NE-sympathetic system with CRH. There is also parallel regulation of both of the central components of the stress system via both stimulatory and inhibitory neurotransmitters and modulators2-4. Glucocorticoids released by the adrenal cortex in response to ACTH exert negative feedback effects, not only on the pituitary ACTH secretion but also on the hypothalamic CRH neuron and the LC/NE-sympathetic system. The second most important modulator of pituitary ACTH secretion is AVP. In addition to the role of magnocellurar AVP neurons in conserving body fluids and controlling plasma osmolality, AVP produced by parvicellular PVN neurons exerts an important role in response to most stress modalities. Vasopressin synergizes with CRH during stress and their secretion is augmented reciprocally, resulting in stimulation and secretion of abundant quantities of ACTH from the pituitary and subsequently cortisol from the adrenals5. CRH appears to play a permissive role in ACTH secretion, whereas AVP and other factors, such as angiotensin II, have synergistic or additive effects. The terminals of the parvicellular PVN CRH and AVP neurons, project not only to the hypophyseal portal system, but to various other sites, including the noradrenergic neurons of the brain stem and the proopiomelanocortin (POMC)-containing neurons in the arcuate nucleus of the hypothalamus. The anterior pituitary is the second level of the HPA axis. The pituitary corticotroph cells synthesize and release ACTH, after stimulation by CRH. The adrenal cortex is the principal target organ of the ACTH and the third level of the HPA axis. The binding of ACTH to high-affinity membrane receptors on the adrenal cells results in the biosynthesis of cortisol. The production of cortisol follows within minutes the ACTH circadian secretion. Cortisol also exerts fast (inhibiting ACTH secretion) and delayed (suppressing CRH and ACTH production) negative feedback actions on its own secretion.
2.2. The HPA axis and the role of proinflammatory cytokines
The stress system is active when the body is at rest, responding to many distinct circadian, neurosensory, blood-borne, and limbic signals6. These signals include cytokines produced by immune-mediated inflammatory reactions, such as tumor necrosis factor-α (TNFα), interleukin 1 (IL-1), and interleukin 6 (IL-6). Immune cells carry receptors for a number of hormones, neuropeptides, and neurotransmitters. During inflammatory stress, the three proinflammatory cytokines TNFa, IL-1, IL-6, stimulate hypothalamic CRH and/or AVP secretion leading indirectly, through glucocorticoid secretion, to limitation of the inflammatory reaction. Thus, there is a reciprocal interaction between the brain - which, by regulating endocrine and peripheral nervous system functions, influences in a major way the immune/inflammatory (I/I) response and thus the defense of the organism - and the products of the I/I reaction which influence the brain along with its endocrine and peripheral nervous system limbs7. The three proinflammatory cytokines are secreted in a cascade-like fashion, with TNFα appearing first, IL-1 second, and IL-6 last. All three cytokines stimulate their own secretion, in an autocrine fashion, from the cells that produce them. Tumor necrosis factor-α and IL-1 stimulate each other's secretion and both promote the release of IL-6. Systemic IL-6 concentrations likewise increase during stress unrelated to inflammation, presumably stimulated by catecholamines through β2-adrenergic receptors. The three proinflammatory cytokines activate the HPA axis independently as well as in combination.
Corticotropin-releasing-hormone-neutralizing antibodies, glucocorticoids, and prostanoid-synthesis inhibitors block the activation of the axis. As we have shown in the past, in humans, IL-6 increases plasma concentrations of ACTH and cortisol well above the concentrations achieved with maximal stimulating doses of CRH8 (Figure 2). On the first day of subcutaneous IL-6 administration to human subjects, the plasma concentrations of ACTH achieved were well above those observed after ovine (o) CRH stimulation, during maximal treadmill exercise, or while undergoing major surgery. The concentration of cortisol was also elevated in a marked and prolonged fashion on the first day. The ACTH response to IL-6 was significantly lower on the 7th than on the 1st day of administration. However, the overall cortisol response on the 7th day was of similar magnitude. The high concentrations of cortisol on the 7th day after IL-6 administration suggest that the HPA axis has reached a new equilibrium at this time. We hypothesize that the glucorticoid negative feedback blunts the ACTH responsiveness to IL-6; the latter, however, sustains the axis activated during the period of its administration. Such a "resetting" of the HPA axis has been previously observed in melancholic depression and the syndrome of late-onset congenital adrenal hyperplasia. Lending further credence to this idea were the adrenal computed tomographic scan findings of one of the patients who had this test performed before and 3 days after the end of the injections. This patient's adrenals were grossly enlarged after the 7th day of IL-6 administration8. An alternative explanation is that chronically administered IL-6 may itself stimulate adrenal cortisol secretion in concert or in synergy with pituitary ACTH, thus participating directly in the observed resetting of the adrenal cortex. Later Spath-Schwalbe et al confirmed the activation of the HPA axis from IL-6 and in parallel showed that administration or elevated levels of this cytokine can cause elevation of the body temperature in the absence of any infectious agent9. We have subsequently shown that, in addition to causing CRH secretion, IL-6 stimulates the other major ACTH segretagogue, AVP, presumably at the parvicellular neuron level at low doses, and clearly at the magnocellular neuron level at high doses10. In addition, it might exert its own ACTH secretagogue effects directly on the pituitary corticotroph. The AVP values correlated significantly with the amount of IL-6 injected, indicating that the latter has a direct and/or indirect dose-dependent stimulatory effect on systemic secretion of AVP via magnocellular neurons (Figure 1). This unequivocal effect of IL-6 suggests that this cytokine may be the mediator through which AVP is stimulated in the syndrome of inappropriate antidiuretic hormone (SIADH) secretion observed in patients with active infectious or inflammatory diseases11.

Figure 2. Mean±SE plasma immunoreactive CRH (top), immunoreactive ACTH (middle), and immunoreactive cortisol (bottom) following a sc bolus injection of IL-6 in cancer patients after the first (circles) and the seventh (triangles) daily IL-6 injection of each cycle of IL-6 administration. The shaded areas represent the mean±SD hormone responses of healthy volunteers to a standard bolus dose (1ìg/kgr) of oCRH.
Furthermore, we have shown that endogenous IL-6 can stimulate the HPA axis under non experimental conditions, in untreated rheumatoid arthritis (RA) patients early in the course of their disease12. Interleukin-6 levels were significantly increased in the RA patients compared to controls during the late afternoon and early morning hours. When time-lagged cross-correlation analysis was performed, a significant positive correlation was observed between IL-6 and ACTH at +60 min for RA patients with IL-6 leading ACTH, followed by a stronger correlation between IL-6 levels and cortisol secretion at +120 min in the same patients with IL-6 leading cortisol. These findings suggest that endogenous IL-6 may directly and/or indirectly stimulate pituitary ACTH and adrenal cortisol production in RA patients. On the other hand, the presence of the negative correlation between IL-6 and cortisol peaking at lagtime-300 min, with IL-6 following cortisol, may indicate the inhibitory effect of cortisol on peripheral production of IL-6. Thus, plasma IL-6 is elevated and displays circadian variation in patients with early untreated RA and it seems likely to stimulate secretion of ACTH and cortisol in these patients.
How inflammatory cytokines reach the hypothalamic CRH and AVP neurons is unclear, given that the blood-brain barrier protects the cell bodies of both kinds of neurons. The proinflammatory cytokines may cause endothelial and glial cells to secrete IL-6 and other mediators of inflammation, which reach the CRH and AVP neurons in a cascade-like fashion13. Alternatively, there may be a special transport system for proinflammatory cytokines, or they may directly activate the terminals of the CRH and AVP neurons in the median eminence, which is outside the blood-brain barrier. Inflammation may also indirectly activate the HPA axis. This could occur through the stimulation of the central noradrenergic stress system by cytokines and other mediators that act first on stress system neurons outside the blood-brain barrier (the area postrema) or on neurons inside the barrier through the endothelial-glial-neuronal cascade mentioned above. In addition to their short-term effects on the hypothalamus, the proinflammatory cytokines can apparently stimulate pituitary ACTH and adrenal cortisol secretion directly at high concentrations or if given adequate time for interaction with these tissues. Normally, the anterior pituitary and adrenal glands produce IL-1 and IL-6, which may influence local hormone production14.
On the other hand, glucocorticoids have antiiflammatory and immunosuppressive properties. They inhibit the production of all three proinflammatory cytokines and also inhibit their effects on target tissues. Cathecholamines, the other end products of the stress system, have a major role in the control of inflammation through stimulation of IL-6, which in its turn inhibits the other two cytokines, stimulates glucocorticoids, and induces the acute-phase response. It is noteworthy that catecholamines and glucocorticoids, stress, and stress hormones influence the development, course, and pathology of certain allergic, autoimmune/inflammatory, infectious, and neoplastic diseases as they favor the TH2 response profile15.
3. EXERCISE AS A STRESS MODEL
3.1. Animals
Exercise as a stress model has been studied in animals. Plasma levels of corticosterone (B) are increased after acute exercise in all mammals that have been studied. Glucocorticoids play an important role in the mobilization of energy reserves during physical activity by stimulating gluconeogenesis, promoting lipolysis and increasing protein catabolism. A wide range of studies in several species of mammals have reported acute increases in the plasma concentration of glucocorticoids in response to submaximal, maximal, and supramaximal exercise. The effect of chronic exercise on the glucocorticoid response is less clear.
In addition to supporting activity via metabolic modulation, glucocorticoids also have complex effects on the central nervous system, including effects on locomotor behavior. In rats, B is necessary for the occurrence of schedule-induced wheel running after adrenalectomy, and chronic B administration to intact animals results in increased locomotor activity. Both the acute and chronic effects of voluntary activity on B secretion were examined in a recent study16. The authors employed a novel animal model, i.e. mice selectively bred for high voluntary wheel running. Female mice were housed with (wheel-access) or without (no-wheel) wheels for 8 wk beginning at 26 days of age. When adjusted for running in the previous 20 min, no difference between wheel-access and no-wheel animals was found. Thus, no training effect on B response was observed. These results constitute strong evidence that in some animals the acute B response is unaffected by chronic voluntary exercise. The high-baseline B levels observed in no-wheel animals may be reflective of a modulation of the HPA axis as a result of selection per increased wheel-running behavior. One potential mediator of this HPA upregulation is CRH. The latter stimulates ACTH and glucocorticoid secretion but also has behavioral effects on the central nervous system17, irrespective of its action on the anterior pituitary. Another explanation could be that for animals selectively bred for high wheel-running activity, the absence of a wheel is psychologically stressful. Furthermore, centrally administered CRH decreased food intake and body mass in rats. By analogy, hypersecretion of CRH may also be responsible for weight loss in anorexia nervosa. In another study, voluntary exercise in male mice did not alter body weights as compared with control animals but there was less abdominal fat, lighter thymuses, and heavier adrenal glands in exercised mice. Body composition was changed in exercising mice, mainly because of substantial loss of abdominal adipose tissue (a result of enhanced lipolysis), skeletal muscle enlargements, and increases in heart weight. Exercise also resulted in asymmetric structural changes in the adrenal glands. In exercising mice, early-morning baseline plasma ACTH levels were decreased as compared to controls, while B levels were not different between the two groups. However, plasma B levels at the start of the dark phase (i.e. 1800) were twice as high as those in control animals. It is well known that the sympathetic activity of the adrenal medulla is a positive modulator of adrenocortical sensitivity to ACTH18. Therefore, the changes that exercise induced in the adrenal glands, i.e. the augmented adrenocortical responsiveness to ACTH, may be caused by the enhanced sympathoadrenomedullary activity resulting in the doubled plasma B levels in the face of unchanged ACTH levels.
In reaction to forced swimming and restraint stress, exercising mice responded with higher B levels than those of the control animals but with similar ACTH levels. Furthermore, CRH mRNA levels in the PVN were lower in the exercising mice. The ACTH data show that despite lower PVN CRH mRNA levels, exercising animals can apparently produce a normal ACTH response to such potentially life-threatening situations. The negative feedback efficacy seems to be normal in exercising mice given the normal peak responses in ACTH and the unchanged glucocorticoid receptor capacity in these animals. The higher glucocorticoid response in exercising mice to forced swimming and restraint corresponds with the enlarged adrenal cortex and the enhanced sympathoadrenomedullary activation. The latter is a response to stressors demanding physical activity, and/or to psychological stress. However, if exposed to a novel environment, exercising mice presented decreased ACTH responses by comparison with those in the sedentary mice, clearly indicating that the exposure to novelty has a lower impact on these animals. This observation is consistent with studies reporting that exercised mice and humans show reduced anxiety, which is in agreement with the lower CRH mRNA levels in the PVN in these animals.
Several studies have also investigated the role of cytokines in exercise and the effects of training on immunity competence. A recent study in pigs indicated that physical stress resulted in immediate increase in the plasma concentrations of cortisol, lactate, and hypoxanthine, but had no effect on the blastogenic capability of lymphocytes or on IL-2 production. The ability of blood mononuclear cells to produce interferon-a in vitro was not affected by exercise stress19. Several reports indicate that exercise elevates plasma levels of proinflammatory cytokines, primarily IL-6. The latter has been shown to be a potent inducer of acute phase protein synthesis in both rats and humans and might be involved in the generation of acute phase responses observed after exercise. To clarify the role of the central and peripheral catecholaminergic system during exercise, rats were injected either intraperitoneally or intracerebroventricularly with 6-hydroxydopamine which depletes the central and peripheral neuron system of catecholamines20. Exercise-induced plasma ACTH, IL-6, and B did not increase after pretreatment with 6-hydroxydopamine. These results suggest that central and peripheral cathecholamines are involved in the regulation of the exercise-induced IL-6 elevation.
3.2. Humans
3.2.1. Exercise and Hypercortisolemia: The role of CRH and AVP
Exercise represents a stress condition since it diverts many systems of the organism—mainly the cardiopulmonary system—to adapt to a new condition. In fact, the two chief stress-activated systems, the LC/NE-sympathetic system (with its endocrine component, the adrenal medulla) and the HPA axis, are activated by exercise and participate in the maintenance of homeostasis and the development of physical fitness.
It has been suggested convincingly that exercise activates the HPA axis by demonstrating that exercise induces an increase of plasma immunoreactive β-endorphin/β-lipotropin in normal volunteers21. In this study, adult male volunteers of similar physical fitness and body composition tested under three exercise-thermoregulatory stress conditions, showed a gradual increase in the concentration of peripheral β-endorphin. Adrenocorticotropin hormone and β-endorphin, both POMC-derived molecules, are secreted in equimolar amounts by the corticotroph cells in the pituitary. In another study, plasma ACTH, cortisol, and lactate responses were measured in sedentary subjects as well as moderately and highly trained runners before and after graded levels of treadmill exercise (50, 70 and 90 percent of maximal oxygen uptake)22. Basal evening concentrations of ACTH and cortisol, but not of lactate, were found elevated in highly trained runners as compared with sedentary subjects and moderately trained runners. Exercise-stimulated ACTH, cortisol, and lactate responses were similar in all groups and were proportional to the exercise intensity employed. These responses, however, were attenuated in the trained subjects when plotted against applied absolute workload. After injection of a bolus oCRH dose, the highly trained group had diminished ACTH and cortisol responses. At the highest relative intensity of treadmill exercise, plasma ACTH concentrations are higher than during the oCRH-stimulation test. In fact, the observed levels are similar to those that have been observed during insulin-tolerance tests and surgery. Highly trained runners demonstrate attenuated responses to exercise as compared to the other two groups, indicating that while these subjects in basal conditions are under mild hypercortisolism, during exercise their HPA axis is "trained" to milder responses. Hence, the activation that acute exercise induces in the HPA axis is inversely proportional to the level of physical training. Daily strenuous exercise appears to lead to chronic ACTH hypersecretion and adrenal hyperfunction, a phenomenon also observed in laboratory animals in which regular strenuous exercise was associated with adrenal hypertrophy. The ability of training to increase the capacity to handle a higher workload with less pituitary-adrenal activation is one of multiple interrelated adaptations and is proportional to the degree of physical training.
The findings of mild evening hypercortisolism and diminished responses of ACTH and cortisol to oCRH in highly trained subjects are similar to observations in patients with anorexia nervosa or depression. In these disorders, CRH hypersecretion has been postulated, both to mediate the accompanying hypercortisolism and to be involved in the development of the symptom complex. This accords with the suggestion that compulsive running is an analogue of anorexia nervosa; it remains to be established whether these findings are specific for runners or apply equally to other types of participants in sports once a certain level of training is attained. Second, highly trained subjects, as well as anorectic and depressed patients, may represent subjects in whom different stressors (excessive running, perceived environmental changes, weight loss, or emotional stress) may have exceeded the putative threshold leading to sustained hypercortisolism.
In conclusion, stimulation of CRH secretion, which leads to ACTH and cortisol hypersecretion, is highly likely during exercise, but this hormone is only secreted in the inaccessible hypophyseal portal system and is rapidly degradated at the level of hypophysis. For that reason, CRH cannot be measured in the systemic circulation and no direct evidence of such hypersecretion has yet been obtained. Plasma AVP concentrations, conversely, are clearly stimulated by exercise in an intensity-dependent fashion. Vasopressin secretion is proportional to the exercise-induced increase in plasma osmolality, a known stimulus of this hormone. Furthermore, blood concentrations of ACTH induced by exoge
nous CRH are less than those observed during stressful conditions, such as exercise, surgery, or hypoglycemia. This suggests that other substances may modulate ACTH release in humans. In a recent study, exercise was used as a physiological stimulus to determine whether CRH is the sole mediator of ACTH release in humans23. To control for the effects of CRH, oCRH was administered at a rate that would saturate the capacity of the corticotroph to respond further to exogenous or endogenous CRH. The results indicated that there was further elevation of plasma ACTH levels during exercise despite elevated preexercise cortisol levels during the CRH infusion. Plasma cortisol levels also rose, but much less impressively than plasma ACTH levels. It was concluded that the enhancement of the ACTH response observed in the subjects receiving oCRH during exercise is caused by other substances which participate in the stimulation of the ACTH release. The less impressive change in the cortisol concentration from pre- to post-exercise during the CRH infusion, compared with that of the ACTH concentration is perhaps because the adrenal cortex had already reached its maximal secretory capacity. Despite the high cortisol levels, postexercise plasma ACTH concentrations during the oCRH infusion increased 3.7-fold over pre-exercise values. Normally, cortisol feedback inhibits ACTH release. This elevation of ACTH in the face of maximal cortisol levels suggests either that the stress of exercise overrides cortisol inhibition of the HPA axis or that other factors, such as AVP, induce ACTH release in synergy with or in addition to CRH and are minimally influenced by negative feedback from cortisol. Indeed, AVP has been shown to synergize CRH-stimulated ACTH secretion through potentiation of the ability of CRH to increase cAMP concentrations in the dispersed rat corticotrophs24. Therefore, AVP might also participate in the ACTH response to exercise. It has been shown that exercise stimulates AVP release into systemic circulation in a manner proportional to exercise intensity25. Angiotensin-II and lactate are two other substances which are stimulated by exercise in proportion to the workload, in a fashion similar to that observed with ACTH, cortisol and AVP25,26.
3.2.2. Exercise and proinflammatory cytokines
In 1996 Papanicolaou et al investigated whether physical stress, such as high-intensity exercise, is related to IL-6 secretion in humans, and to what extent this is correlated with catecholamines secretion27. In experimental animals, stress and catecholamines stimulate endogenous IL-6 secretion, whereas glucocorticoid inhibits it. Catecholamines stimulate IL-6 secretion via β-adrenergic receptor28. Fifteen male volunteers were submitted to high-intensity treadmill exercise test runs in a double-blind crossover design, and plasma IL-6 and cathecholamines concentrations were measured before and after exercise tests, after pretreatment with placebo, hydrocortisone (HC), or dexamethasone (DEX). Plasma epinephrine and noreepinephrine concentrations peaked 15 min after the start of exercise, whereas plasma IL-6 concentrations peaked twice, 15 min, and 45 min after the onset of the test run. There was no difference in either epinephrine or norepinephrine peaks among the three treatments, but the net area under the curve for IL-6 was smaller after hydrocortisone or dexamethasone than after placebo, and smaller after dexamethasone than after hydrocortisone. A positive correlation was observed between peak plasma epinephrine or norepinephrine and IL-6 at 15 min. Exercise increases the plasma concentration of catecholamines in humans in an intensity-dependent fashion. In this study, although pretreatment with either glucocorticoid resulted in lower basal norepinephrine than those after placebo, there was no apparent effect of either glucocorticoid on the exercise-induced increase in plasma levels of either catecholamine. In other words, glucocorticoids do not inhibit the sympathoneural discharge in response to acute stimuli. Plasma IL-6 concentrations also rose in response to exercise. This increase can be due to increased production and/or decreased clearance of IL-6 during exercise. The rapid elevation of plasma IL-6 concentrations strongly supports the former mechanism. Pretreatment with either glucocorticoid, HC, or DEX, resulted in a significant attenuation of the IL-6 response to exercise without abolishing it. This is in accordance with the findings of other studies, where glucocorticoids inhibit the production of IL-6 in vitro and in vivo in animals and humans29. The IL-6 response to surgical trauma was attenuated by exogenous glucocorticoids, further supporting the idea that glucocorticoids inhibit, without completely abolishing, the secretion of IL-6 during stressful conditions30. The difference in the net AUC for IL-6 observed with regard to the two glucocorticoids administered in this study is probably due to differences in their potencies in this particular system. On the basis of animal and human studies the correlation observed between peak plasma catecholamines and peak plasma IL-6 levels may represent a cause-and-effect phenomenon. IL-6 is produced by a variety of cells, but primarily by monocytes/macrophages, endothelial cells, fibroblasts, lymphocytes, and mast cells31. It is therefore possible that sympathetic activation stimulates IL-6 production and/or secretion in two ways: First, in a paracrine fashion, through norepinephrine released at the sympathetic nerve terminals innervating immune organs (such as the spleen and the thymus) containing IL-6 producing cells; and second, in an endocrine fashion, primarily through epinephrine secretion from the adrenal medulla and the distant effects of this hormone on IL-6 producing cells. Thus, high-intensity exercise stimulates the secretion of IL-6, possibly by catecholamines, whereas exogenous glucocorticoids attenuate this effect without affecting the catecholamine levels. One might suggest that exercise resembles aseptical inflammation inducing IL-6 production, which is inhibited by anti-inflammatory substances such as glucocorticoids.
To examine both whether exercise-induced IL-6 secretion is also responsible for part of the HPA axis stimulation during exercise, as well as to study the response-capacity of this axis in an autoimmune disease, 37 patients with sarcoidosis (27 untreated and 10 treated with corticosteroids) and 13 normal healthy subjects were submitted to maximal cardiopulmonary exercise testing (CPET) on a treadmill32. Cortisol and IL-6 were measured in plasma samples drawn before, at peak, and 15 min after (recovery) CPET. Baseline cortisol levels in plasma were within normal range and comparable in sarcoidosis patients with and without treatment (Figure 3) as well as in healthy volunteers (Figure 4). Baseline serum IL-6 levels were significantly elevated in all37 patients with sarcoidosis (Figure 3) compared to healthy volunteers (Figure 4). Sarcoidosis is a delayed type hypersensitivity form of granulomatous disease, suggesting a type 1 cytokine response33 and characterized by normal or increased levels of interferon-γ, IL-2, IL-12 and TNF-β. These cytokines are involved in the pathogenesis of autoimmune diseases, such as experimental autoimmune uveitis, experimental allergic encephalomyelitis, or insulin-dependent diabetes mellitus34. On the other hand, the type 2 cytokine response is characterized by an impaired cellular response with an increase in one or more B-cell activities and increased levels of IL-4, IL-5, IL-6, IL-10, and/or IL-13. These cytokines exert a down-regulatory activity on an exaggerated and/or inappropriate type cytokine response33. Thus, the increased circulating IL-6 levels in sarcoidosis patients might reflect the activation of type 2 mechanism. The significant increase of baseline serum IL-6 levels in patients with sarcoidosis was not paralleled by an increase of basal cortisol levels, despite the fact that IL-6 is a major stimulating factor of the HPA axis. Baseline serum TNF-α levels were elevated in patients with sarcoidosis compared to healthy subjects, whereas baseline serum IL-6 levels were comparable between these groups. During exercise and recovery, cortisol levels were significantly elevated in patients without treatment and in healthy subjects as compared to baseline. This elevation parallels the significant elevation of the IL-6. Interleukin-6 is a key mediator in the activation of the HPA axis during exercise since the exercise-induced increase of endogenous IL-6 in normal subjects leads to stimulation of the HPA axis. This exercise-induced increase of IL-6 is probably norepinephrine- and epinephrine-mediated27. Thus, in patients with sarcoidosis without treatment, exercise is still a potent stimulator of the HPA axis via the IL-6 elevation even though IL-6 levels at baseline are already significantly elevated in these patients as compared to healthy volunteers. However, cortisol to IL-6 ratio in sarcoidosis patients without treatment is significantly lower than that of healthy subjects at the peak of exercise and during recovery. Therefore, even though IL-6 remains a potent stimulator of the HPA axis for these patients, the secreted cortisol levels are lower than in healthy subjects for equivalent amounts of IL-6, reflecting a dysfunction of the HPA axis response to IL-6 in sarcoidosis. Conversely, in treated sarcoidosis patients, IL-6 levels increased during exercise and recovery whereas cortisol levels did not, indicating that chronic treatment with increased doses of corticosteroids impairs the IL-6-induced HPA axis stimulation probably at the hypothalamic level. Thus, in patients with sarcoidosis the "inappropriately" normal cortisol levels at baseline in face of increased IL-6 levels together with the lower cortisol to IL-6 ratio after exercise indicate a defect of the HPA. The "inappropriately" normal cortisol levels could participate in the pathogenesis of sarcoidosis by facilitating the expression of T helper (TH) 1 autoimmune phenomena.
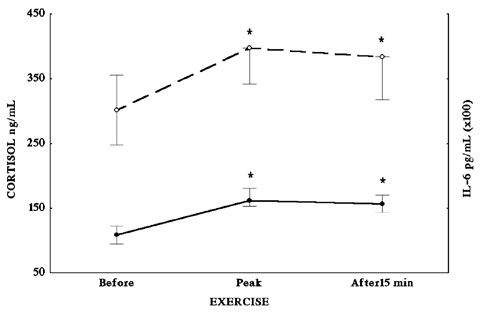
Figure 3. Cortisol (solid line) and IL-6 (dotted line) plasma levels in patients with sarcoidosis at baseline, during and 15 min after treadmill exercise.

Figure 4. Cortisol (solid line) and IL-6 (dotted line) plasma levels of healthy volunteers at baseline, during and 15 min after treadmill exercise.
3.2.3. Exercise and effects on immunity
If high-intensity exercise is viewed as a stress situation, it might well influence the type of the immune response. Stress situations could influence the expression of TH1 and TH2 response profiles15. Epidemiological and experimental studies suggest that stress and stress-related hormones (glucocorticoids, epinephrine, norepinephrine) influence the development, course, and pathology of certain allergic, autoimmune/inflammatory, infectious, and neoplastic diseases by predominately stimulating a TH2 as opposed to a TH1 type response, that is, by enhancing humoral more than cellular immunity35. The stress-induced dominance of TH2 type immune responses results from the control of specific key regulatory cytokines such as interferon-γ, IL-4, IL-10, and IL-1235. Both IL-12 and interferon-γ stimulate the development of the TH1 phenotype while suppressing the expression of the TH2 phenotype. Conversely, IL-4 stimulates the development of TH2 cells but suppresses TH1 differentiation from uncommitted CD4+ cells. Interleukin-10, a product of TH2 cells and monocytes/macrophages, is also a potent inhibitor of TH1 cytokine secretion. Both cAMP elevation and glucocorticoids (35, 36) are more potent inhibitors of IL-2 and interferon-γ by TH1 cells than IL-4 secretion by TH2 cells, favoring a TH2 shift. The bulk of evidence indicates that glucocorticoids favor TH2 immune cells and mediators associated with humoral immunity over those linked to cellular immune responses. Similarly, substances that increase cAMP including adrenergic agents are more potent inhibitors of TH1 cells responses, consequently favoring TH2 responses. Recently it has been demonstrated37 that dexamethasone, norepinephrine, and epinephrine inhibited lipopolysaccharide-induced IL-12 production by human peripheral blood mononuclear leukocytes ex vivo. This cytokine is known to favor TH1 type of immunity. These results provide strong evidence that stress hormones selectively amplify TH2 responses while suppressing TH1 by altering the production of key regulatory cytokines farther upstream of the cytokine cascade. Chronic strenuous-exercise leads to increased basal glucocorticoid levels as well as catecholamines levels during exercise which could favor TH2 immunity profile.
3.2.4. Exercise and other hormones (Growth Hormone and Prolactin)
Besides activation of the HPA axis, IL-6 is known to stimulate two other hormones: growth hormone (GH) and prolactin (PRL), which on the evolutionary scale are also related to stress response. In the study reported above, Luger et al examined, apart from the HPA axis, the effect of acute exercise on plasma GH and PRL concentrations in three groups of healthy male volunteers38. Acute treadmill exercise induced elevations of plasma GH and PRL concentrations in all subjects tested. There was an intensity-dependent stimulation of PRL, i.e. no response to exercise at a relative intensity of 50% VO2max, a significant response at 70% VO2max, and an even greater response at 90% VO2max. In contrast, a significant GH response to exercise was obtained already at 50% VO2max, with a maximal stimulation at 70% VO2max, which was not different from the one observed at 90% VO2max. These observations indicate that several different mechanisms and/or mediators are involved in the exercise-induced hormonal changes, or that the threshold sensitivity to exercise is different in these different hormone systems. One of these might be the exercise-induced increase in body core temperature. It is interesting that GH response to exercise tends to be lower than that observed after exogenous administration of GH releasing hormone (GHRH), the hypothalamic releasing factor for GH, suggesting stimulation via an alternate pathway or simultaneous release of an inhibiting factor, possibly somatostatin. The latter is possible since CRH, which is expected to rise in exercise, has been shown to stimulate somatostatin release. Also, the PRL response to exercise was smaller when compared with other established releasers, such as TRH or metoclopramide. Chronic physical training leads to adaptive changes in GH and PRL responses. Thus, hormonal responses to absolute workloads were inversely proportional to the degree of training, i.e. at a workload where the GH and PRL response was already maximal in untrained subjects, no response could be registered in the highly trained subjects. However, when corrected for the training level, by expressing the workload as percentage of the individual maximal workload (VO2max), similar GH and PRL responses occurred at similar relative work intensities in the three groups of subjects tested. An alternative mechanism of PRL release in exercise attributes a role to lactate or other metabolic factors. To test this hypothesis, lactate was infused intravenously in normal healthy volunteers. With an infusion of 1M sodium lactate, lactate concentrations in plasma ranged between those observed at an exercise intensity of 70 and 90% VO2max. Although this was followed by a significant rise in plasma PRL and GH concentrations, the increases observed were clearly smaller than those following exercise at intensities causing similar plasma lactate concentrations. It seems therefore, as previously suggested, that lactate may play some, but not an exclusive, role in the exercise-induced release of GH and PRL. This view is corroborated by the fact that GH release was also observed when exercise was perfomed at intensity below the anaerobic threshold (i.e. at 50% VO2max), at which no significant lactate production was detected. Because of the low exercise intensity needed to obtain a maximal GH response, exercise can be regarded as a suitable stimulation test for GH. Activation of GH and PRL release occurs at different exercise intensities, GH being more sensitive than PRL. In other words, during exercise, besides CRH, ACTH, AVP and cortisol, PRL and GH are possibly stimulated by cytokines in response to stress.
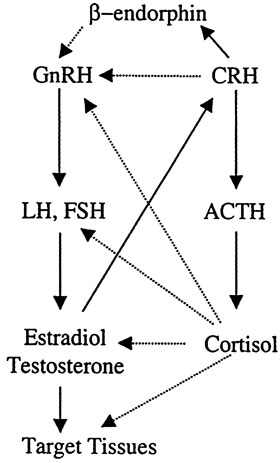
Figure 5. Heuristic representation of the interactions between the hypothalamic-pituitary-adrenal axis and the hypothalamic-pituitary-gonadal axis. Activation of the stress system leads to inhibition of the reproductive axis by direct or indirect inhibition of the GnRH neuron by CRH, beta endorphin, and glucocorticoids.
To investigate whether IL-6 might be a mediator to GH and PRL release, Tsigos et al perfomed dose-response studies of recombinant human IL-6 on pituitary hormone secretion in 15 healthy male volunteers39. By measuring resting metabolic rate (RMR) with indirect calorimetry and plasma anterior pituitary hormones, they found that IL-6 acutely causes dramatic increases in GH secretion in man. Maximum GH release was 100-fold that of baseline. Interestingly, stimulation of GH release occurred even with such doses of IL-6 that did not appreciably influence HPA axis activity or thermogenesis. This suggests a higher sensitivity of the GH axis to IL-6 and possibly to the other proinflammatory cytokines, similar to that of acute-phase reactant production. Prolactin showed a similar but less pronounced response pattern. As in the case of ACTH secretion, the acute effects of IL-6 on GH and PRL release are also thought to be mediated via actions of this cytokine on hypothalamic control centers40. Direct actions of IL-6 on the pituitary to modify anterior pituitary hormone release do appear to exist but are unlikely to have contributed to the observed responses as they usually take several hours (more than 4-8 h) to appear40. Thus, one can suggest that the GH and PRL elevations observed during exercise are caused directly and/or indirectly by IL-6. Bremer et al. compared women with psychogenic hypothalamic amenorrhea to women with normal menstrual cycle during exercise, they found that in the former PRL did not increase whereas in the latter PRL and GH did so. Apparently in the group with the psychogenic hypothalamic amenorrhea, the defect is located at the level of hypothalamus or higher and the specific regulatory neurons cannot be stimulated by the exercise-induced cytokines. In contrast, women with normal menstrual cycle have an integral hypothalamus which can be stimulated by IL-6 during exercise41.
3.2.5. Exercise and the reproductive system
The hypothalamic-pituitary-gonadal (reproductive) axis is inhibited at all levels by various components of the HPA axis (Figure 5). Either directly or through arcuate POMC neuron β-endorphin release, CRH suppresses the gonadotropin-releasing hormone (GnRH) neurons of the arcuate and preoptic nuclei. Glucocorticoids exert inhibitory effects at the level of GnRH neuron, the pituitary gonadotroph, inhibiting primarily the secretion of luteinizing hormone (LH) as well as the hormonal secretion of the gonads and rendering target tissues of sex steroids resistant to these hormones. Corticotropin-releasing hormone is also a potent negative regulator of LH effects in Leydig cells. This inhibitory loop serves to attenuate androgen production by LH42. It is well known that stress is accompanied by both an increase in the activity of the HPA axis and a decrease in reproductive functions in order to preserve the organism's state of alert at the expense of gonadal activity in case of emergency. In animals, the relationship between population density and reproductive efficiency has been studied in wild rodents5. This effect of the HPA axis on gonadal function depends on the type, duration, and frequency of the stimulus. Specifically, there is evidence that the response of the pituitary-testicular system to aversive stimuli is potentially biphasic, with an initial stimulatory phase and, if stress is prolonged or of sufficient magnitude, a subsequent inhibitory phase. There is also strong evidence that the immediate changes in LH secretion are mediated at the level of GnRH-secreting neurons, while long-term effects also involve peripheral mechanisms such as alteration in pituitary and gonadal responsiveness43.
During inflammatory stress, the inflammatory cytokines suppress reproductive function at several levels. These effects are exerted directly and indirectly by activating hypothalamic neural circuits that secrete CRH and POMC-derived peptides, as well as by peripheral elevations of glucocorticoids44. Suppression of gonadal function caused by chronic HPA axis activation has been demonstrated in highly trained runners of both sexes, in ballet dancers, and in individuals sustaining anorexia nervosa or starvation. Chronic amenorrhoea of obligate athletes that train hard should be mentioned here5. These subjects demonstrate a relative baseline hypercortisolism, with males exhibiting low LH and testosterone levels and females presenting hypogonadotropic hypogonadism and amenorrhea. The stress of exercise acts upon the HPA axis and the LC/NE-sympathetic system leading to production of glucocorticoids and catecholamines and, through them as well as through the CRH neuron, inhibits the gonadal function (Figure 6). Strenuous exercise can also affect the age of menarche if it is initiated before menarche. Among ballet dancers who start training before their menarche, it has been shown that the more they dance per week the more their puberty is delayed and the more commonly they suffer amenorrhea45.
4. SHORT- AND LONG-TERM CONSEQUENCES
There has been a substantial increase in women participating in sports over the last 30 years. While exercise provides many health benefits, there appears to be a unique set of risks associated with intense exercise for the female athlete. The so-called "female athlete triad" encompasses the risks of amenorrhea, osteoporosis, and eating disorders, three distinct medical conditions that may occur in girls and women athletes46. Other investigators speak of "exercise-related female reproductive dysfunction" (ERFRD), which can possibly include short-term (infertility) and long-term (osteoporosis) consequences47.
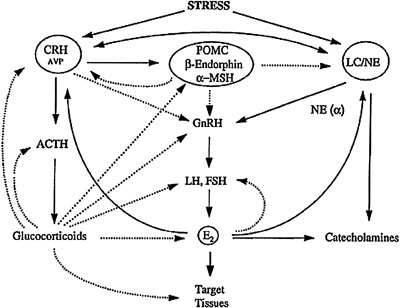
Figure 6. Heuristic representation of the interplay among the hypothalamic-pituitary-adrenal axis, the locus ceruleus/norepinephrine (LC/NE) sympathetic system and the hypothalamic-pituitary-gonadal axis. The dotted lines represent inhibition while the solid lines represent stimulation.
4.1. Short-term consequences
Clinical or biochemical abnormalities of gonadal function, consisting of delayed puberty/menarche, luteal phase deficiency, oligo-amenorrhea, or anovulation occur in girls and women participating in strenuous sports. Indeed, the prevalence of "athletic amenorrhea" is 4-20 times higher than that in the general population48. Depending on the time that strenuous exercise begins, it can cause delayed menarche (15,1 ± 0.5 yr) if it starts before puberty, or menstrual cycle irregularities if it starts after puberty. Menstrual abnormalities in the female athlete result from the suppression of the spontaneous pulsatile hypothalamic secretion of GnRH, which results in decreased pulsatile secretion of LH and FSH and shuts down the stimulation of the ovary. A number of factors, such as energy balance, exercise intensity, and training practices, bodyweight and fat composition, disordered eating behaviors, and physical and emotional stress levels, may contribute to the development of athletic menstrual dysfunction49. The stress of exercise, the concomitant activation of the HPA axis, and the observed hypercortisolemia are some of the main factors that lead to the suppression of the HPG axis. It was also proposed that the disturbance of the GnRH oscillator is caused by either an insufficient estrogen or progesterone feedback, or by an imbalance of local opioid peptide and catecholamine activities mediated by GABA, CRH and insulin-like growth factor (IGF) 150. Recent studies have suggested that reduced energy availability (increased energy expenditure with inadequate caloric intake) is another of the main causes of the central suppression of the HPG axis. The role of low energy availability on both the development and the reversal of exercise-induced amenorrhea were studied in a longitudinal study using a monkey model. The connecting signal between metabolic status and reproductive dysfunction may be represented by a substance such as insulin, amino acids, IGF binding protein 1, leptin, and cortisol51. In other words, the observed hypoinsulinemia in amenorrheic athletes may provide a mechanism through which adipose tissue detects energy deficiency and, in turn, downregulates leptin (ob) gene expression and secretion. The hypoleptinemia that is also observed is consistent with the hypothesis that the dependence of reproductive function on energy availability may be mediated by leptin.
4.2. Long-term consequences
Long-term strenuous exercise has been considered to lead to osteopenia/osteoporosis, increased risk for coronary heart disease, and the euthyroid "sick" syndrome. Osteoporosis can occur at any age. In the premenopausal stage, full achievement of peak bone mass may be curtailed and accelerated bone loss may occur in early adulthood. Exercise can be one among several factors causing pre
menopausal osteoporosis52. Indeed, strenuous exercise causes hypercortisolism, hypogonadism, and nutritional depletion, which includes inadequate calcium and vitamin D intake53. Adolescence is a period of rapid skeletal growth during which nearly half of adult skeletal mass is acquired. This life stage is a window of opportunity for influencing peak bone mass and augmenting or reducing the risk of osteoporosis later in life. Endocrine factors that may influence peak bone mass include IGF-1 which regulates skeletal growth, and sex steroids which stimulate epiphyseal maturation. All these parameters are diminished in athletes54. Besides this particular period of life, throughout life the adult skeleton undergoes a continuous turnover, termed bone remodeling, during which old bone is resorbed by osteoclasts and new bone is formed by osteoblasts on surfaces on which resorption has recently been completed. Bone homeostasis depends on the orderly replenishment of cellular constituents and its destruction leads to osteoporosis, which represents aberrant cell production relative to demand55. The increase in IL-6 secretion during exercise may participate in the pathogenesis of the osteoporosis observed in obligate athletes by stimulating osteoclastic activity. Indeed, by stimulating the development of osteoclasts, IL-6 play a profound role in skeletal homeostasis while IL-6 may promote the development of osteoblasts. Moreover, it is now established that bone-active systemic hormones, such as sex steroids, parathyroid hormone, parathyroid hormone-related peptide, 1, 25-dihydroxyvitamin D3, and thyroxin, exert their potent influences on bone remodeling and skeletal homeostasis by regulating the production and action of IL-6 and IL-1. Hormones affect the actions of these interleukins by regulating the expression of the receptors of these cytokines56. On the other hand, cortisol, which is elevated in athletes, strongly inhibits the osteoblastic activity. Recent studies have in fact shown that the effects of glucocorticoids on bone are dependent on the autocrine actions of the enzyme 11beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1). Expression of 11 beta-HSD1 in osteoblasts facilitates local synthesis of active glucocorticoids with consequent effects on osteoblastic proliferation and differentiation. Specifically, glucocorticoids decrease the replication of cells of the osteoblastic lineage and the function of the osteoblast. Moreover, it has been found that within bone cells inflammatory cytokines, which in general instigate tissue damage, cause an autocrine switch in intracellular corticosteroid metabolism by disabling glucocorticoid inactivation (11 beta-HSD2) while inducing glucocorticoid activation (11 beta-HSD1)57. In addition, recent studies have shown that, gonadal steroids inhibit the osteoclasts, IGF-1 inhibits the osteoclasts after energy deficit, and GH increases the osteoblastic activity during energy deficit. All these factors are low in athletes58. Hence athletes, and especially female athletes, are at risk for decreased bone mineral density and present increased risk for developing osteoporosis and stress fractures.
Premenopausal reduction in endogenous estrogen are hypothesized to be similarly pathogenic and to accelerate development of coronary heart disease59. These functional abnormalities, which occur along a continuum between mild, estrogen deficiency luteal phase progesterone deficiency, and amenorrhea, are relatively common and are often attributed to exercise, psychological stress, and energy imbalance. These deficits can accelerate the development of cardiovascular disease. Further more exercise-related hypercortisolemia can cause endothelial dysfunction, which aggravates atherosclerosis and ultimately increases the risk of coronary heart disease.
Lastly, the third long-term consequence of exercise is euthyroid "sick" syndrome (ESS) that occurs during acute critical nonthyroidal illnesses and is characterized by abnormalities of thyroid function and possibly reflect a stress situation. The most common abnormalities are low serum total triiodothyronin T3 and increased serum levels of reverse T3 (rT3)60 and increased levels of IL-6, TNFa, and cortisol. Currently much attention is being centered on the role of IL-6 and TNFa in developing ESS through their effect on the HPA axis and especially on glucocorticoids release61. The glucocorticoids released during stress suppress the thyroid axis function in many ways; they suppress TSH secretion, decrease T3 formation from tetraiodothyronine (T4) in peripheral tissues and increase the production of rT3. Moreover, they inhibit type 5'-deiodinase activity. Exercise represents a stress condition duringwhich hypercortisolemia occurs as well as stimulation of endogenous IL-6 secretion and can thus cause ESS. Furthermore, if exercise-related energy expenditure exceeds the calories consumed, a condition of ESS may be induced. In female athletes, 4 days of low energy availability led to reduced T3 and, free T3, increased rT3, and slightly increased T462. Since an adequate amount of the prohormone T4 was available throughout the study, an alteration in the peripheral metabolism of T4 was hypothesized. The increase in rT3 and decrease in T3 were consistent with a decreased activity 5'-deiodinase activity since this enzyme is responsible for the production of T3 and the clearance of rT3.63
REFERENCES
1. Chrousos GP, 1992 Regulation and dysregulation of the hypothalamic-pituitary-adrenal axis. Endocrinol Metab Clin North Am 21: 833-858.
2. Mastorakos G, Ilias I, 2003 Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann NYAcad Sci 997: 1-14.
3. Tsigos C, Chrousos GP, 1994 Physiology of the hypothalamic-pituitary-adrenal axis in health and dysregulation in psychiatric and autoimmune disorders. Endocrinol Metab Clin North Am 23: 451-466.
4. Larsen PJ, Jessop D, Patel H, Lightman SL, Chowdrey HS, 1993 Substance P inhibits the release of anterior pituitary adrenocorticotrophin via a central mechanism involving corticotrophin-releasing factor-containing neurons in the hypoyhalamic paraventricular nucleus. J Neuroendocrinol 5: 99-105.
5. Kamal EH, Gold PW, Chrousos GP, 2001 Neuroendocrinology of stress. Endocrinol Metab Clin North Am 30: 695-728.
6. Chrousos GP, 2004 The hypothalamic-piyuitary-adrenal axis and immune-mediated inflammation. New Engl J Med 332: 1351-1362.
7. Mastorakos G, Bamberger C, Chrousos GP 1999 Neuroendocrine regulation of the immune process. In: Plotnikoff NP (ed) Cytokines-Stress and Immunity, Modern Endocrinology and Diabetes, CRC Press LLC, USA, 17-37.
8. Mastorakos G, Chrousos GP, Weber JS, 1993 Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab 77: 1690-1694.
9. Spath-Schwalbe E, Born J, Schrezenmeier H, et al, 1994 Interleukin-6 stimulates the hypothalamus-pituitary-adrenocortical axis in man. J Clin Endocrinol Metab 79: 1212-1214.
10. Mastorakos G, Weber JS, Magiakou MA, Gunn H, Chrousos GP, 1994 Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion. J Clin Endocrinol Metab 79: 934-939.
11. Gionis D, Ilias I, Moustaki M, et al, 2003 Hypothalamic-pituitary-adrenal axis and interleukin-6 activity in children with head trauma and syndrome of inappropriate secretion of antidiuretic hormone. J Ped Endocrinol Metab 16: 49-54.
12. Crofford LJ, Kalogeras KT, Mastorakos G, et al, 1997 Circadian relationships between interleukin-6 (IL-6) and hypothalamic pituitary-adrenal axis hormones: Failure of IL-6 to cause sustained hypercortisolism in patients with early untreated rheumatoid arthritis. J Clin Endocrinol Metab 82: 1279-1283.
13. Tilders FJ, DeRijk RH, Van Dam AM, et al, 1994 Activation of the hypothalamic-pituitary-adrenal axis by bacterial endotoxins: routes and intermediate signals. Psychoneuroendocrinology 19: 209-232.
14. Imura H, Fucata J, Mori T, 1991 Cytokines and endocrine functions: an interaction between the immune and neuroendocrine systems. Clin Endocrinol 35: 107-15.
15. Webster EL, Torpy DJ, Elenkov IJ, Chrousos GP, 1998 Corticotropin-releasing hormone and inflammation. Ann N Y Acad Sci 840: 21-32.
16. Girad I, Garland TJr, 2002 Plasma corticosterone response to acute and chronic voluntary exercise in female house mice. J Appl Physiol 92: 1553-1561.
17. Owens MJ, Nemeroff CB, 1991 Physiology and pharmacology of corticotrophin-releasing factor. Pharmacol Rev 43: 425-473.
18. Rhodes JS, Hosasack GR, Girard I, et al, 2001 Differential sensitivity to acute administration of cocaine, GBR 12909, and fluoxetine in mice selectively bred for hyperactive wheel-running behavior. Psychopharmacology 158: 120-131.
19. Waern MJ, Fossum C, 1993 Effects of acute physical stress on immune competence in pig. Am J Vet Res 54: 596-601.
20. Yu XN, Komaki G, Sudo N, Kubo C, 2001 Central and peripheral catecholamines regulate the exercise-induced elevation of plasma interleukin 6 in rats. Life Sci 69: 167-174.
21. Kelso TB, Herbert WG, Gwazdauskas FC, Goss FL, Hess JL, 1984 Exercise-thermoregulatory stress and increased plasma β-endorphin/β-lipotropin in humans. J Appl Physiol 57: 444-449.
22. Luger A, Deuster PA, Kyle SB, et al, 1987 Acute hypothalamic-pituitary-adrenal responses to the stress of treadmill exercise. N Engl J Med 316: 1309-15.
23. Smoak B, Deuster P, Rabin D, Chrousos GP, 1991 Corticotropin-releasing hormone is not the sole factor mediating exercise-induced adrenocorticotropin release in humans. J Clin Endocrinol Metab 73: 302-306.
24. Abou-Samra A-B, Harwood JP, Manganiello VC, Catt KJ, Aguilera G, 1987 Phorbol 12-myristate 13-acetate and vasopressin potentiate the effect of corticotrophin-releasing factor on cyclic AMP production in rat anterior piyuitary cells. J Biol Chem 262: 1129-36.
25. Covertino VA, Keil LC, Bernauer EM, 1981 Plasma volume, osmolality, vasopressin, and rennin activity during graded exercise in man. J Appl Physiol 50: 123-128.
26. Deuster PA, Chrousos GP, Luger A, et al, 1989 Hormonal and metabolic responses of untrained, moderately trained, and highly trained men to three exercise intensities. Metabolism 38: 141-148.
27. Papanicolaou DA, Petrides JS, Tsigos C, et al, 1996 Exercise stimulates interleukin-6 secretion: inhibition by glucocorticoids and correlation with cathecholamines. Am J Physiol 271: E601-E605.
28. DeRijk RH, Boelen A, Tilders FJH, Berkenbosch F, 1994 Induction of plasma interleukin-6 by circulating adrenaline in the rat. Psychoneuroendocrinology 19: 155-163.
29. Rock CS, Coyle MS, Keogh CV, et al, 1992 Influence of hypercortisolemia on the acute-phase protein response to endotoxin in humans. Surgery 112: 467-474.
30. Hogevold HE, Kierulf P, Ovstebo R, Reikeros O, 1992 Acute phase reactants and interleukin6 after total hip replacement; effects of high dose cortcosteroids. Eur. J Surg 158: 339-345.
31. Terebuh PD, Otterness IG, Strieter RM, et al, 1992 Biologic and immunohistohemichal analysis of interleukin-6 expression in vivo. Constitutive and induced expression in murine polymorphonuclear and mononuclear phagocytes. Am J Pathol 140: 649-657.
32. Mastorakos G, Nanas S, Pappa E, et al, 1998 Interleukin 6 changes during cardiopulmonary exercise test in sarcoidosis patients and controls. 80th Annual meeting of the Endocrine Society, New Orleans, Louisiana.
33. Lucey DR, Clerici M, Shearer GM, 1996 Type1 and type 2 cytokine dysregulation in humans infectious, neoplastic, and inflammatory disease. Clin Microbiol Rev 9: 532-562.
34. Singh VK, Mehrotra S, Agarwal SS 1999 The paradigm of Th1 and Th2 cytokines: its relevance to autoimmunity and allergy. Immunol Res 20: 147-161.
35. Wilder RL, 1995 Neuroedocrine-immune system interactions and auto-immunity. Annu Rev Immunol 13: 138-146.
36. Ramirez F, Fowell DJ, Puklavec M, Simmonds S, Mason D 1996 Glucocorticoids promote a TH2 cytokine respose by CD4+ T cells in vitro. J Immunol 156: 2406-2412.
37. Elenkov IJ, Papanicolaou DA, Wilder RA, Chrousos GP, 1996 Modulatory effects of glucocorticoids and cathecholamines on human interleukin-12 and intrleukin-10 production: clinical implications. Proc Assoc Am Physicians 108: 374-381.
38. Luger A, Watschinger B Deuster P, et al, 1992 Plasma growth hormone and prolactin responses to graded levels of acute exercise and to a lactate infusion. Neuroendocrinology 56: 112-117.
39. Tsigos C, Papanicolaou DA, Defensor R, et al, 1997 Dose effects of recombinant human interleukin-6 on pituitary hormone secretion and energy expenditure. Neuroendocrinology 66 54-62.
40. McCann SM, Karanth S, Kamat A, et al, 1994 Induction by cytokines of the pattern of pituitary hormone secretion in infection. Neuroimmunomodulation 1: 2-13.
41. Bremer B, Cumming D, Stern B, et al, 1984 Hormonal responses to acute exercise in women with psychogenic hypothalamic amenorrhea and exercise-associated amenorrhea. Abstract #281, 31st Annual Meeting of the Society for Gynecologic Investigation, San Francisco, CA.
42. Ulisse S, Fabbri A, Dufau M, 1989 Corticotropin-releasing factor receptors and actions in rat leydig cells. J Biol Chem 264: 2156-2163.
43. Rivier C, Rivest S, 1991 Effect of stress on the activity of the hypothalamic-pituitary-gonadal axis: peripheral and central mechanisms. Biol Reprod 45: 523-532.
44. Elenkov IJ, Webster EL, Torpy DJ, et al, 1999 Stress, corticotrophin-releasing hormone, glucocorticoids, and the immune/inflammatory response: Acute and chronic effects. Ann N Y Acad Sci 876: 1-13.
45. Frisch RE, Gotz-Welbergen AV, McArthur JW, et al, 1981 Delayed menarche and amenorrhea of college athletes in relation to age of onset of training. JAMA 246: 1559-1563.
46. Papanek PE, 2003 The female athlete triad: an emerging role for physical therapy. J Orthop Sports Phys Ther 33: 594-614.
47. Cannavo S, Curto L, Trimarchi F, 2001 Exercise-related female reproductive dysfunction. J Endocrinol Invest 24: 823-832.
48. Eliakim A, Beyth Y, 2003 Exercise training, menstrual irregularities and bone development in children and adolescents. J Pediatr Adolesc Gynecol 16: 201-206.
49. Manore MM, 2002 Dietary recommendations and athletic menstrual dysfunction. Sports Med. 32: 887-901.
50. DeCree C, 1998 Sex steroid metabolism and menstrual irregularities in the exercising female. A review.
Sports Med 25: 369-406.
51. Mocanu V, Luca VC, Stoica AR, Zbranca E, 2001 The influence of body weight upon the function of ovarianaxis. Rev Med Chir Soc Med Nat Iasi 105: 469-474.
52. Gourlay ML, Brown SA, 2004 Clinical considerations in premenopausal osteoporosis. Arch Intern Med 164: 603-614.
53. Jacoangeli F, Zoli A, Taranto A, et al, 2002 Osteoporosis and anorexia nervosa: relative role of endocrine alterations and malnutrition. Eat Weight Disord 7: 190-195.
54. Weaver CM, 2002 Adolescence: the period of dramatic bone growth. Endocrine 17: 43-48.
55. Papanicolaou DA, 1998 The pathophysiologic roles of interleukin-6 in humans disease. Ann Intern Med 128: 127-137.
56. Manolagas SC, Jilka RL, Belido T, O'Brein CA, Parfitt AM, 1996 Interleukin-6 type cytokines and their receptors in: Bilezikian JP, Raisz LG, Rodan GA, (eds) Principles of Bone Biology, San Diego, Academic Pr; pp, 701-713.
57. Cooper MS, Bujalska I, Rabbitt E, et al, 2001 Modulation of 11beta-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine swith from glucocorticoid inactivation to activation. J Bone Miner Res 16: 1037-1044.
58. Weiler U, Finsler S, Claus R, 2003 Influence of cortisol, gonadal steroids and an energy deficit of biochemical indicators of bone turnover in Swine. J Vet Med A Physiol Pathol Clin Med 50: 79-87.
59. Kaplan JR, Manusk SB, 2004 Ovarian dysfunction, stress, and disease: a primate continuum. ILAR J. 45: 89-115.
60. Chopra IJ, 1997 Euthyroid sick syndrome: is it a misnomer? J Clin Endocrinol Metab 82: 329-334.
61. Michalaki M, Vagenakis AG, Makri M, Kalfarentzos F, Kyriazopoulou V, 2001 Dissociation of the early decline in serum T3 concetration and serum IL-6 rise and TNFa in nonthyroidal illness syndrome induced by abdominal surgery. J Clin Endocrinol Metab 86: 4198-4205.
62. Loucks AB, Callister R, 1993 Induction and prevention of low-T3 syndrome in exercising women. Am J Physiol 264: R924-R930.
63. Loucks AB, Heath EM, 1994 Induction of low-T3 syndrome in exercising women occurs at a threshold of energy availability. Am J Physiol 266: R817-R823.
Address correspondence and requests for reprints to:
George Mastorakos, 3 Neofytou Vamva Str.,
10674, Athens, GREECE, Fax: 30-210-3636229,
e-mail: mastorak@mail.kapatel.gr
Received 23-12-04, Revised 08-03-05, Accepted 15-03-05