Department of Endocrinology, University of Munich, Germany
OBJECTIVE: As we previously demonstrated, the inhibitory effect of iodine on thyroid cell growth is mediated by iodolactones, especially 6-iodo-5-hydroxy-eicosatrienoic acid (δ-iodolactone). In this communication we compare the effect of iodide, molecular iodine and δ-iodolactone on growth inhibition and apoptosis on three human thyroid carcinoma cell lines (B-CPAP cells, FTC-133 cells and 8505C cells) as well as on human breast cancer cells (MCF 7). METHODS: Thyroid carcinoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and MCF 7 cells in Rowswell Park Memorial Institute (RPMI) culture medium, both containing 10% (v/v) Fetal Calf Serum (FCS), until they were confluent. Around 2000 cells were then distributed in 12-well plates and grown for 48 h in either DMEM (thyroid cancer cells) or in RPMI medium (MCF 7 cells) both containing 5% FCS. Thereafter, different concentrations of iodide, iodine or δ-iodolactone were added for 24 h. Growth rate was estimated by cell counting in a Coulter Counter adapted for epithelial cells. Apoptosis was determined by a mitochondrial potential assay. RESULTS: The growth rate of B-CPAP cells was unaffected by iodide, but was reduced by high concentreations of molecular iodine (100 and 500 μM). However, δ-iodolactone significantly reduced cell proliferation already with low concentrations (5 μM and 10 μM) and further in a dose-dependent manner up to 82%. FTC-133 and 8505C cells were unaffected by iodide, iodine or δ-iodolactone. In contrast, in MCF 7 cells, molecular iodine (100 μM) inhibited growth from 100% to 83% but δ-iodolactone (1, 5 and 10 μM) dose-dependently decreased growth rate from 100% to 82% and 62%, respectively. The inhibition of growth was through apoptosis, and not necrosis, as the amount of apoptotic cells corresponded to the growth inhibition. CONCLUSION: δ-Ιodolactone seems to be the main iodocompound which can inhibit growth and induce apoptosis in B-CPAP cells as well as in MCF 7 breast cancer cells.
Apoptosis, Iodide, δ-Iodolactone, Mammary carcinoma, Molecular iodine, Thyroid carcinoma
INTRODUCTION
Although iodocompounds other than thyroid hormones were detected in thyroid homogenates, and iodolipids have been detected after radioiodine incorporation studies since the early fifties,1 their physiological role was unknown. It was hypothesized, however, that these substances are involved in thyroid autoregulation.2
Specific compounds were identified when new, highly sensitive methods, such as gas Chromatography-Mass Spectrometry (GC-MS), were used.3 The in-vitro synthesis of β-iodolactone by incubating arachidonic acid with lactoperoxidase and H2O2 was first described by Boeynam in rat thyroid slices.4 The main product of the conversion of arachidonic acid was identified as 6-iodo-5-hydroxy-8,11,14-eicosatrienoic acid β-lactone (Figure 1) and was later identified by ex vivo studies in porcine thyroid follicles as well as by in vivo experiments in human thyroid tissue derived from patients treated with high doses of iodine before surgery.3,5 δ-Ιodolactone can be purified by silica gel chromatography and characterized by GC-MS spectroscopy.3 The effect of β-iodolactone has been further investigated in isolated porcine thyroid follicles ex vivo. δ-Iodolactone has no effect on cAMP formation in porcine thyroid follicles and seems to be exclusively involved in cAMP-independent growth control. It was shown that β-iodolactone inhibited Epidermal Growth Factor (EGF)-induced proliferation of isolated porcine follicles dose-dependently, comparably to iodide but at about 50-fold lower concentrations.5 Also, in FRTL-5 cells β-iodolactone inhibits thyroid cells proliferation.7 δ-Ιodolactone inhibits inositol-1,4,5-triphosphate (IP3) formation induced by EGF but not by TSH induced IP3 formation. Therefore, β-iodolactone has been postulated to act as a specific inhibitory mediator of iodide on growth factor-induced thyroid cell proliferation.8
Iodolactone ring formation can also be generated with eicosapentaenoic acid or docosahexaenoic acid in vitro using the same method as described for arachidonic acid. The β-iodolactone derived from eicosapentaenoic acid is more active in growth inhibition than β-iodolactone from arachidonic acid, but the β-iodolactone from docosahexaenoic acid was ineffective.9 This indicates the specificity of different iodolactones and might explain why fish and iodide consumption is important for maintaining normal thyroid volume.
During goitrogenesis, endothelial growth precedes the proliferation of thyroid cells. Thyroid follicles ex vivo also produce a Paracrine Endothelial Growth Factor (FGF1) which stimulates fibrobast and endothelial growth. This is inhibited by both iodide and β-iodolactone.10 These findings explain the inhibitory effect of iodide on goitrogenesis, which obviously is also mediated by β-iodolactone. The expression of Transforming Growth Factor beta 1 (TGFβ1) mRNA is unaffected by iodide and β-iodolactone, indicating that TGFβ1 is not involved in the inhibitory effect of iodine or β-iodolactone on thyroid cell growth.11
We have also shown that both iodide and β-iodolactone induce apoptosis, demonstrated by electron microscopy in porcine thyroid follicles ex vivo in a three-dimensional tissue culture.12 Apoptosis was already induced by 0.05 µM of β-iodolactone, whereas 20 µM of iodide were necessary to obtain a comparable effect. The effect of iodide was inhibited by methimazole in contrast to β-iodolactone, indicating that peroxidase activity is necessary for the action of iodide on growth and apoptosis.
Interestingly, the induction of apoptosis was lowered by preincubating human thyroid follicles with low concentrations of selenium (10 and 100 nM), which induced glutathionperoxidase activity. This likely indicates that the induction of apoptosis is mediated by free oxygen radicals in mitochondria.13
Recently it has been shown for the first time that molecular iodine is incorporated in MCF-7 cells independently of Sodium-Iodide-Symporter (NIS) and inhibits proliferation in a comparable manner to β-iodolactone. Uptake of molecular iodine is saturable, suggesting that molecular iodine is able to iodinate arachidonic acid within the cell membrane.14 These cells express lactoperoxidase and therefore exhibit the comparable machinery to metabolise iodine to β-iodolactones like normal thyroid cells. This is a new and exciting field with possible implications for the role of iodine and β-iodolactone in the treatment of breast cancer.15,16
We have now investigated the effect of iodide, iodine and β-iodolactone on malignant thyroid cells in vitro and compare this effect to that of β-iodolactone on malignant breast cancer cells (MCF-7). These cells do not trap iodide, but they may respond to β-iodolactone possibly through a direct effect on cell growth, independent of iodide uptake.
MATERIAL AND METHODS
Materials and cell culture
Dulbecco’s modified Eagle’s medium (DMEM), RPMI-1640, Hank’s balanced salt solution, fetal bovine serum (FBS), streptomycin, penicillin, amphothericin B and trysin-EDTA were obtained by GIBCO-BRL/ invitrogen (Karlsruhe, Germany); soybean trypsin inhibitor was purchased from Calbiochem (La Jolla, USA); potassium iodide and H2O2 were from Merck (Darmstadt, Germany); the Mitochondrial Potential Assay was from R&D Systems (Minneapolis, USA), and all other chemicals were from SIGMA Chemicals Co. Human breast cancer cells were from ATCC HTP-22, while human malignant thyroid cells (B-CPAP cells derived from papillary thyroid carcinoma, FTC-133 derived from follicular thyroid carcinoma and 8505C derived from anaplastic thyroid carcinoma) were kindly provided by Dr. Huang-Vu, University of Halle, Germany. The thyroid carcinoma cells were grown in DMEM with 10% (v/v) FBS, 100 U/ml penicillin, 100 µg/ml streptomycin and 0.5 µg/ml amphothericin B. MCF 7 cells were cultured in RPMI containing 10% (v/v) FCS and antibiotics until they were confluent. Then around 2000 cells were distributed in 12-well plates and grown for 48 h in RPMI containing 5% FCS. Thereafter, different concentrations of iodide, iodine (Lugol’s solution: I2 + I-→ I3-+ I2 → I5-) or β-iodolactone were added for another 24 h. Cells were counted in a Coulter-Counter adapted to the different cell sizes.
δ-Ιodolactone (6-iodo-5-hydroxy-8,11,14-eicosatrienoic acid) was synthesized by lactoperoxidase catalyzed iodination of arachidonic acid and purified by reverse-phase HPLC according to the methods described earlier.3
The mitochondrial potential assay from R&D Systems was used for the detection of apoptosis as recently described.13 The follicles were stained in the well using DePsipher (5, 5’6, 6’-tetrachloro-1, 1’, 3, 3’-tetraethylbenzimidazolyl carbocyanine iodide), a mitochondrial activity marker. The substance accumulates in the intact mitochondrium in the form of aggregates, displaying a bright red signal under a fluorescein filter (495 nm). After the breakdown of the mitochondrial membrane potential that occurs in the early phase of apoptosis, the dye remains in the cytosol as a monomer, causing a green fluorescence.
The cells were counted independently by two experienced technicians. At least 500 thyroid follicular cells of each well were evaluated by each technician and the results were expressed as mean. As a positive control, apoptosis was induced in HL-60 cells with etoposide.13
Statistical analysis
Each experiment with six parallel samples was carried out at least four times independently. The rate of apoptosis was expressed as percentage. Consequently, the samples were regarded as dependent and differences were calculated by Wilcoxon anlysis for paired samples. Statistical calculation was performed with the commercial SPSS program.
RESULTS
Thyroid carcinoma cells
Neither iodide, Lugol’s solution (mainly iodine) (100 µM) nor β-iodolactone (5 and 10 µM) had any effect on cell growth of 8505C cells derived from anaplastic thyroid tissue, nor on FTC-133 cells derived from a metastasis of a follicular carcinoma (data not shown).
In contrast, growth of B-CPAP cells derived from papillary thyroid carcinoma significantly and dose-dependently were inhibited by β-iodolactone (9.3% with 5 µM, p = 0.04, 17.4% with 10 µM, p=0.006) (Figure 2). Whereas iodide, even in high concentrations up to 500 µM, had no effect on the growth of B-CPAP cells, 100 µM and 500 µM of iodine significantly inhibited cell proliferation (Figure 3).
MCF-7 breast cancer cells
Iodide in concentrations of 1, 10 and 100 µM had no significant effect on growth of MCF 7 cells. In contrast, 100 µM iodine significantly inhibited growth from 100% to 85% (p<0.05) (Figure 4). Growth inhibition by β-iodolactone was dose-dependent and already significant with 1 µM of β-iodolactone (Figure 5). With 10 µM β-iodolactone the growth inhibition was 47%. This growth inhibition was due to apoptosis, and not necrosis, as the amount of apoptotic cells corresponded with the growth inhibition (Figure 6). Up to 43% of MCF-7 cells underwent apoptosis after 24-hour incubation with 10 µM β-iodolactone. Moreover, with iodide and more so with iodine, the rate of apoptotic cells was increased, which, however, was not reflected in the determination of cell count as a parameter for growth (Figure 6).
DISCUSSSION
Whereas the role of iodide and β-iodolactone on growth inhibition and induction of apoptosis in normal thyroid follicular cells is well established,8,12 no data are currently available for human thyroid carcinoma cells. Iodide as well as iodine had no effect on growth of carcinoma cells derived from anaplastic and follicular carcinoma. However, there was a mild, but significant, inhibition in the proliferation of papillary thyroid carcinoma cells (B-CPAP) with high doses of iodine. With regard to β-iodolactone, effectiveness was demonstrated already in low concentratons. This finding is in contrast to human breast cancer cells (MCF 7), which respond to iodine, as well as to β-iodolactone with growth inhibition and induction of apoptosis. Apoptosis seems to be the main mechanism of growth inhibition, as the number of apoptotic cells corresponds to the degree of growth inhibition. This has already been demonstrated in normal porcine thyroid follicles ex vivo.12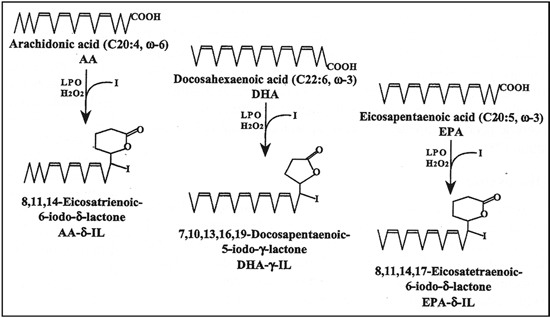
Figure 1. Formula of iodolactones, derived from arachidonic acid, docosahexaenoic acid and eicosapentaenoic acid (adapted from reference 9).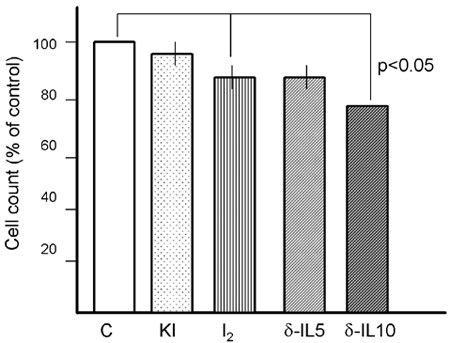
Figure 2. Effect of iodide (KI 100 µM), iodine (100 µM I2) and δ-iodolactone 5 µM (δ-IL5) and 10 µM (δ-IL 10) on B-CPAP thyroid papillary carcinoma cells. Cells were incubated for 24 hours and then cell number counted in a Coulter Counter. Inhibition with 100 µM I2 and 5 and 10 µM of δ-IL was significant (p<0.05), in comparison to controls (C).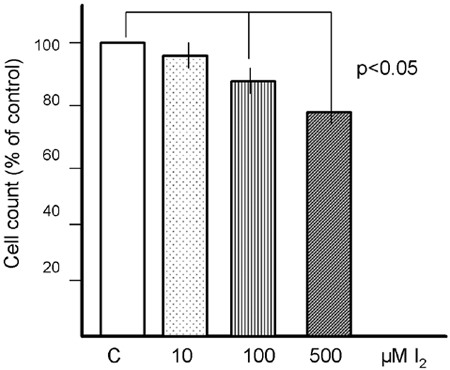
Figure 3. Dose-dependent effect (10, 100 and 500 µM) of iodine (I2) on growth of C-BAP thyroid carcinoma cells. Cells were incubated for 24 hours and then cell number counted in a Coulter Counter. Reduction of cell number with 100 µM and 500 µM I2 was significant (p<0.05), in comparison to controls (C).
The MCF-7 cells express NIS only after incubation with all-trans Retinoic Acid (RA) or dexamethasone.14 In our experiments, NIS was not induced by RA and there was no uptake of iodide (data not shown). Recently, however, it has been shown that MCF-7 cells are able to bind molecular iodine in a saturable manner.14 It is suggested that iodine in the presence of peroxidases within or at the cell membrane might generate β-iodolactones, which then exert their inhibitory activity.15 Interstingly, molecular iodine, but not iodide, inhibits chemically induced mammary carcinogenesis in animals.16 It can be concluded from our experiments that β-iodolactone is the main component generated during iodine exposure of cells expressing peroxidases, which then inhibits growth and induces apoptosis.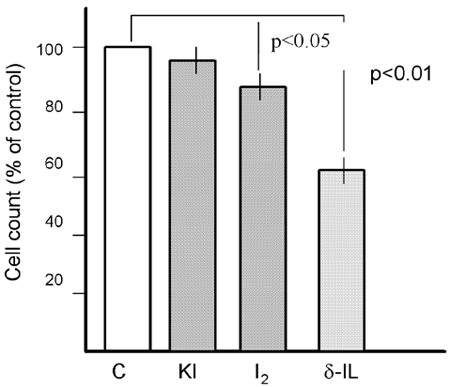
Figure 4. Effect of iodide (KI 100 µM), iodine (100 µM) and δ-iodolactone 5 µM (δ-IL5) and 10 µM (δ-IL 10) on MCF-7 mammary carcinoma cells. Cells were incubated for 24 hours and then cell number counted in a Coulter Counter. Inhibition with 100 µM I2 and 5 µM δ-iodolactone was significant (p<0.01), in comparison to controls (C).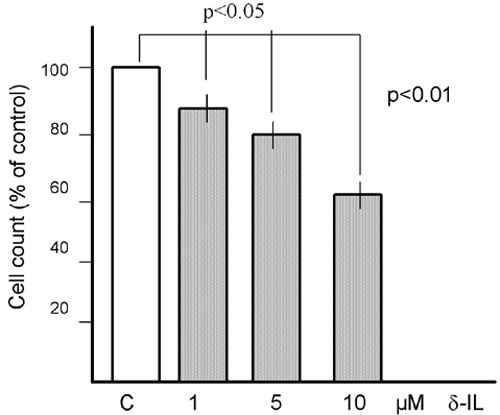
Figure 5. Dose-dependent effect of 1, 5 and 10 µM of δ-iodolactone (δ-IL) on growth of MCF-7 mammary carcinoma cells. Cells were incubated for 24 hours and then cell number counted in a Coulter Counter. Reduction of cell number was already significant with 1 µM δ-iodolactone (p<0.05) and dose-dependently with higher concntrations (p<0.01), in comparison to controls (C).
We had demonstrated earlier that β-iodolactone inhibits IP3 generation but not cAMP formation in porcine thyroid follicles ex vivo and suggested that the inhibition of the protein kinase C pathway might be responsible for the growth inhibition.6 Iodine as well as β-iodolactone also seems to induce apoptosis, and this might be the most important activity of iodine and β-iodolactone, whereas β-iodolactone seems to be the main active component.12 It has also been shown that molecular iodine induces caspase-independent apoptosis in human breast carcinoma cells, involving the mitochondria-mediated pathway.21 These experiments need to be done with β-iodolactone for comparison.
Figure 6. Percentage of apoptotic MCF-7 cells induced by 100 µM iodide (KI), 100 µM iodine (I2) and 10 µM δ-iodolactone (δ-IL) and in controls. Apoptosis was determined by DePsipher mitochondrial assay. …. p<0.05; _ _ _ p<0.01.
The fact that thyroid carcinoma cells do not respond to molecular iodine might be due to a lack of peroxidases (TPO) even in differentiated thyroid carcinomas.17 Rediffentiation studies would be necessary either with RA or rosiglitazone.18-20 These rediffentiation experiments as well as chronic treatment with high doses of molecular iodine might prove to be a promising new approach in the management of patients with differentiated thyroid carcinomas, (not trapping iodide) as well as of patients with breast cancer.
In conclusion, based on current findings we postulate that iodide can generate β-iodolactone only in cells expressing NIS and peroxidases, whereas molecular iodine can be generated within the cell membrane in the presence of peroxidases alone and without the need for NIS. β-Iodolactone is the main compound that inhibits cell proliferation and induces apoptosis in differentiated thyroid carcinoma cells as well as in breast cancer cells.
REFERENCES
1. Taurog A, Tong W, Chaikoff IL, 1957 An unidentified iodine compound formed by incubation of cell-free preparations of tissue with iodine 131 I. J Biol Chem 227: 759-772.
2. Wolff J, 1989 In: “Control of the Thyroid Gland” (eds R. Ekholm, L.D. Kohn, S.H. Wollman) pp 211-244 Plenum Publishing Corp.
3. Dugrillon A, Bechtner G, Uedelhoven WM, Weber PC, Gärtner R, 1990 Evidence that an iodolactone mediates the inhibitory effect of iodide on thyroid cell proliferation but not on cyclic AMP formation. Endocrinology 127: 337-343.
4. Boeynaems JM, Hubbard WC, 1980 Transformation of arachidonic acid into an iodolactone by the rat thyroid. J Biol Chem 255: 9001-9004.
5. Dugrillon A, Uedelhoven WM, Pisarev MA, Bechtner G, Gärtner R, 1994 Identification of δ-iodolactone in iodide treated human goiter and its inhibitory effect on proliferation of human thyroid follicles. Horm Metab Res 26: 465-469.
6. Dugrillon A, Gärtner R, 1995 Delta-Iodolactone decrease epidermal growth factor-induced inositol - 1,4,5-triphosphate generation in porcine thyroid follicles - a possible mechanism of growth inhibition by iodide. Europ J Endocrinol 132: 735-743.
7. Pisarev MA, Bocanera LV, Chester HA, et al, 1992 Effect of iodoarachidonates on thyroid FRTL-5 cells growth. Horm Metab Res 24: 558-561.
8. Pisarev MA, Gärtner R, 2000 Thyroid autoregulation by iodine. In “The Thyroid” (ed LE Braverman and RD Utiger) Lippincott Williams & Wilkins, Philadelphia, Baltimore, New York, London, Buenos Aires, Hong Kong, Sidney, Tokyo; pp, 85-90.
9. Gärtner R, Dugrillon A, Bechtner G, 1996 Evidence that thyroid growth autoregulation is mediated by an iodolactone. Acta Med Austriaca 23: 47-51.
10. Greil W, Rafferzeder M, Bechtner G, Gärtner R, 1989 Release of an endothelial cell growth factor from cultured porcine thyroid follicles. Mol Endocrinol 3: 858-867.
11. Gärtner R, Schopohl D, Schaefer S, et al, 1997 Regulation of transforming growth factor ß1 messenger ribonucleic acid expression in porcine thyroid follicles in vitro by growth factors, iodine or δ-iodolactone. Thyroid 7: 633-640.
12. Langer R, Burzler C, Bechtner G, Gärtner R, 2003 Influence of iodide and iodolactones on thyroid apoptosis. Exp Clin Endocrinol Diabetes 111: 325-329.
13. Lehmann P, Rank P, Hallfeldt KL, Krebs B, Gärtner R, 2006 Dose-related influence of sodium selenite on apoptosis in human thyroid follicles in vitro induced by iodine, EGF, TGF-beta, and H2O2. Biol Trace Elem Res 112: 119-130.
14. Arroyo-Helguera O, Anguiano B, Delgado G, Aceves C, 2006 Uptake and antiproliferative effect of molecular iodine in the MCF-7 breast cancer cell line. Endocr Relat Cancer 13: 1147-1158.
15. Aceves C, Anguiano B, Delgado G, 2005 Is iodine a gatekeeper of the integrity of the mammary gland? J Mammary Gland Biol Neoplasia.10: 189-196.
16. García-Solís P, Alfaro Y, Anguiano B, 2005 Inhibition of N-methyl-N-nitrosourea-induced mammary carcinogenesis by molecular iodine (I2) but not by iodide (I-) treatment. Evidence that I2 prevents cancer promotion. Mol Cell Endocrinol 31: 49-57.
17. Faggiano A, Caillou B, Lacroix L, et al, 2007 Functional characterization of human thyroid tissue with immunohistochemistry. Thyroid 17: 203-211.
18. Puppin C, D’Aurizio F, D’Elia AV, et al, 2005 Effects of histone acetylation on sodium iodide symporter promoter and expression of thyroid-specific transcription factors. Endocrinology 146: 3967-3674.
19. Kebebew E, Peng M, Reiff E, et al, 2006 A phase II trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer. Surgery 140: 960-966.
20. Kogai T, Schultz JJ, Johnson LS, Huang M, Brent GA, 2000 Retinoic acid induces sodium/iodide symporter gene expression and radioiodide uptake in the MCF-7 breast cancer cell line. Proc Natl Acad Sci USA 97: 8519-8524.
21. Shrivastava A, Tiwari M, Sinha RA, et al, 2006 Molecular iodine induces caspase-independent apoptosis in human breast carcinoma cells involving the mitochondria-mediated pathway. J Biol Chem 281: 19762-19771.
Address for correspondence:
Prof. Dr. Roland Gärtner, Medizinische Klinik Innenstadt, University of Munich,
Ziemssenstrasse 1, D-80336 Munich, Germany,
Tel: +49 89 5160 2332, Fax: +49 89 5160 2124, e-mail: roland.gaertner@med.uni.muenchen.de
Received 10-04-09, Revised 20-09-09, Accepted 20-10-09