Department of Medical Genetics, University of Athens, “Choremis” Research Laboratory, “Aghia Sophia” Children’s Hospital, Athens, Greece
The end-organ resistance to androgens has been designated as Androgen Insensitivity Syndrome (AIS), an X-linked disorder caused by mutations in the Androgen Receptor (AR) gene. It is generally accepted that defects in the AR gene prevent the normal development of both internal and external genital structures in 46,XY individuals, causing a variety of phenotypes ranging from male infertility to completely normal female external genitalia. Precise diagnosis requires clinical, hormonal and molecular investigation and is of great importance for appropriate gender assignment and management in general. The complexity of phenotypic presentation of AIS with genotype-phenotype variability of identical mutations complicates both the diagnostic procedure and genetic counseling of the affected families. More than 400 different AR gene mutations have thus far been reported but the receptor structure-function relationship and its phenotypic outcome is not yet fully understood. This review focuses on the clinical features and molecular pathophysiology of AIS and explores the relationship of the molecular defects in the AR gene to their clinical expression.
AIS, Ambiguous genitalia, Androgen Receptor Gene, AR, CAIS, Intersex disorders, MAIS, PAIS
INTRODUCTION
Androgen Insensitivity Syndrome (AIS; testicular feminization; OMIM# 300068) is an X-linked disease characterized by variable defects in virilization of 46,XY individuals. This is due to loss-of -function mutations in the androgen receptor gene (AR; OMIM# 313700), which results in peripheral androgen resistance.
Since 1953, when the first description of the AIS was reported by John Morris,1 many crucial steps have been taken in the elucidation of the underlying mechanisms of this disease. In 1970, Lyon and Hawkes2 reported an X-linked gene for testicular feminization (Tfm) in the mouse. Further genetic studies established the X-chromosomal localization of the Tfm locus2,3 and later Migeon et al4 demonstrated the homology of the locus for the human disorder to that in the mouse. In 1989, the exact localization of the human Androgen Receptor gene (AR gene) was defined on Xq11-125 and in the same year the first proof that AIS was caused by mutations in the AR gene was published by Brown et al.6 The final evidence of the molecular background of the AIS was offered by the report on the sequence of the intron-exon boundaries of the human AR gene7 in 1989.
The clinical phenotypes of AIS are variable and are classified into three main categories: complete (CAIS), partial (PAIS) and mild (MAIS) form, the designations reflecting the severity of androgen resistance.
AR GENE
The AR gene is a single copy gene that spans ~90 Kb of genomic DNA and lies on the X-chromosome at Xq11–12.7-9 The protein coding region (~2757 Kp open reading frame) comprises 8 exons, designated A-H7 or 1-8,10 separated by introns up to 26 Kb in size8 (Figure 1). Exon 1 encodes the amino-terminal domain (NTD) which comprises more than half of the AR protein. Toward the 5?end of exon 1 is a CAG repeat region (polyglutamine, PolyQ) that contains an average of 21±2 repeats. This repeat region is highly polymorphic and is found expanded in patients with Kennedy disease11 as well as in infertile men.12 Downstream of the PolyQ region, near the 3?-end of NTD, there is another polymorphic polyglycine (PolyG) region. Exons 2-3 encode the DNA-binding domain, exons 4 (3?-end region) to 8 encode the ligand-binding domain, while the 5?-region of exon 4 encodes the hinge region.13
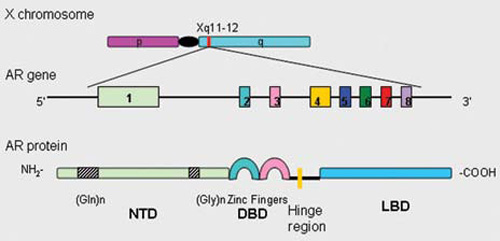
Figure 1. Human androgen receptor gene is mapped to the long arm of the X-chromosome (Xq11-12). The human androgen receptor protein is encoded by 8 exons (1-8). Similarly to other nuclear receptors, the protein consists of several distinct functional domains: the NH2-terminal domain (NTD) containing two polymorphic stretches [(Gln)n and (Gly)n], the DNA-binding domain (DBD), the hinge region and the ligand binding domain (LBD).
ANDROGEN RECEPTOR AND MECHANISMS OF ANDROGEN ACTION
Androgens exert their effects by mediating the differentiation and development of the normal male phenotype via a single receptor protein, the androgen receptor (AR).14 The androgen receptor belongs to the superfamily of nuclear receptors which includes receptors of other steroid hormones.13,15 It is expressed in fetal tissues as early as 8 weeks of gestation, before the onset of androgen action, and is activated in a ligand-dependent manner to coordinate expression of suitably responsive genes. In the human male embryo, testes begin to secrete androgens at 9 weeks of gestation. Endogenous androgens, testosterone (T) and dihydrotestosterone (DHT) form a complex with AR, giving rise to different biological messages.16,17 Testosterone, which peaks between 11 and 18 weeks of gestation, stimulates differentiation of the Wolffian duct system into epididemis, vas deferens and seminal vesicles. Development of the prostate from the urogenital sinus and masculinization of the primordial external genital into penis and scrotum require the more potent androgen, DHT. DHT is derived from testosterone via the action of the enzyme 5alpha-reductase type 2 which is expressed in these tissues. Mediation of the action of both testosterone and DHT requires the presence of AR in target tissues.18
AR protein consists of 919 amino acids, has a molecular mass of 110 kD and is organized in three main structural domains (Figure 1):
- The N-terminal domain (NTD) or transactivation domain is the least homologous in sequence and most variable in size domain among the members of the steroid receptor family. This domain is mainly involved in the regulation of target gene transcription as well as in transcriptional regulation via protein-protein interactions with other transcription factors.19-22
- The DNA binding domain (DBD) is the most highly conserved part of the receptor molecule, which determines the specificity of AR interaction with DNA.23,24 DBD consists of two zinc clusters: one is involved in direct DNA-binding and contains the so-called P-box for the specific recognition of the androgen-response element, while the other is implicated in protein-protein interactions and serves as a stabilization unit for the dimerization of the two receptor molecules.
- The ligand-binding domain (LBD) functions principally by specific, high-affinity binding of androgens. In addition, the LBD is also involved in nuclear localization, receptor dimerization and interaction with other proteins.8 The amino acid sequence of this region displays about 50% homology with the corresponding residues in glucocorticoid, mineralcorticoid and progesterone receptors.25
Between the DBD and the LBD lies the hinge region (encoded by the 5?-region of exon 4), which contains the major part of the AR nuclear targeting signal and mediates the transfer of AR from the cytoplasm to its site of action in the nucleus.19,20,26
Recently, the complex crystal structure of the human androgen receptor LBD and the synthetic ligand methyltrienolone (R1881) were determined by Matias et al.27 Elucidation of the 3-D crystallographic structure established that 12-alpha-helices and 4 short β-strands, associated in 2 anti-parallel β sheets, form a typical helical sandwich. This structure is involved in the construction of the ligand-binding pocket allowing an interaction surface for binding proteins, such as co-activators and co-repressors. The ligand-binding pocket of the androgen receptor ligand-binding domain contains 20 amino acid residues that interact with the bound ligand.27
In the absence of ligand, the AR resides in the cytoplasm.28 Hormone binding induces a transconformation of the receptor and allows its dimerization and translocation into the nucleus, where it initiates transcription through specific interactions with the transcription machinery (Figure 2).29 Any event which could impair the normal function of the AR may, among others, result in insufficient androgen action in a male fetus and undervirilization of the male newborn.30

Figure 2. Ligand-dependent activation of the androgen receptor. Androgens such as DHT diffuse through the plasma membrane and bind to the AR. Upon ligand binding, the AR undergoes conformational changes involving an NH2-/carboxyl-terminal interaction and receptor stabilization. The AR translocates to the nucleus where dimerization and DNA binding to regulatory androgen response elements occurs. AR (androgen receptor); DHT (dihydrotestosterone); CBP (CREB-binding protein); ARE (androgen response element); hsp (heat shock protein); SRC-1 (steroid receptor coactivator 1).29
CLINICAL FEATURES
Androgen insensitivity syndromes probably represent the most common identifiable cause of male pseudohermaphroditism.31,32 Affected individuals usually present with a 46,XY karyotype, incompletely descended testes and female or partially masculinized external genitalia. AIS has traditionally been classified into three clinical subgroups based on the genital phenotype: complete (CAIS), partial (PAIS) and mild or minimal (MAIS) androgen insensitivity syndrome. In table 1 the clinical features of AIS subgroups are summarized.25
Complete Androgen Insensitivity Syndrome (CAIS)
The estimated prevalence of CAIS is about 1:20,000-64,000 male births.25,33 Individuals affected by CAIS present normal female external genitalia. They may present with a short blind ending vagina, absence of Wolffian duct derived structures like epididymides, vas deferens and seminal vesicles, absence of prostate. At puberty breast development is observed but pubic and axillary hair are absent.25 Müllerian structures rarely occur in CAIS individuals.34-36 In the extensive study of Rutgers and Scully, however, Müllerian duct derivatives, such as diminutive fallopian tubes, were detected in up to one third of the 43 cases studied.37 In a study from our group which included 11 patients with CAIS, only one individual, a 14.5-year-old girl, who carried the new mutation p.L881P, exhibited a unilateral fallopian tube structure.38 Wolffian duct derivatives are also rarely found.39 Subjects with CAIS are born unambiguously female and are not suspected of being abnormal until the onset of puberty, when breast development is normal but pubic and axillary hair is not developed and menses do not occur. A number of patients are recognised when they present inguinal hernia. The age at onset of breast development has not been studied extensively but has been reported as delayed in some individuals, being more consistent with the onset of puberty in males.25,40,41 A recent retrospective study of 9 postpubertal individuals with CAIS suggests that these individuals enter puberty at an age closer to that of females.42
Timing of referral to clinicians of individuals affected with CAIS extends from birth or even before birth to adulthood. The reasons for early referral (fetal life or infancy) is a discrepancy between the finding of a 46,XY karyotype on amniocentesis and the presence of female external genitalia on prenatal ultrasound examination or during clinical examination at birth.43,44 Later on, the development of inguinal hernia, often bilateral, raises the question of AIS. Grumbach and Conte45 estimated the prevalence of AIS in phenotypically female infants presenting with inguinal hernia to be 1-2%.
Partial Androgen Insensitivity Syndrome (PAIS)
Partial or incomplete forms of AIS comprise a wide spectrum of clinical phenotypes. Because of variability of clinical manifestations and the existence of subtle or atypical forms of androgen resistance such as male infertility,46-48 the prevalence of partial forms of AIS is unknown. As a group, however, these disorders may be at least as common as the CAIS.
The various phenotypes of PAIS reflect the severity of undervirilization. Individuals with predominantly female external genitalia present mild clitoromegaly, some fusion of the labia and pubic hair at puberty, while those of predominantly male appearance of external genitalia exhibit micropenis, perineal hypospadias and cryptorchidism (also called Reifenstein syndrome, OMIM# 312300).25,49 In the group of PAIS individuals, Wolffian duct derived structures can be partially or fully developed, depending on the biochemical phenotype of the AR, while testes are more frequently found undescended (in the inguinal region or in scrotum/labia majora).50 At puberty, elevated luteinizing hormone, testosterone and estradiol levels are observed, but in general, the degree of undervirilization is less as compared with individuals with CAIS.13,51 Affected subjects usually develop gynecomastia with no significant increase in the size of phallus. The testes have a reduced number of germ cells with consequent azoospermia and they may later on, at puberty, develop in situ carcinoma.50
Mild Androgen Insensitivity Syndrome (MAIS)
MAIS is the phenotype at the other extreme to CAIS. Genitalia may be underdeveloped for a male or there may be simple coronal hypospadias or a prominent midline raphe of the scrotum.50 At puberty, MAIS takes two phenotypic forms, both presenting with various degrees of gynecomastia, high-pitched voice, sparse sexual hair and impotence. In one form of MAIS, spermatogenesis and fertility are impaired,53,54 while in another spermatogenesis is normal or sufficient to preserve fertility.52,55 Although experience with MAIS families is limited, they appear to harbor relatively little phenotypic disparity.
Subjects with MAIS AR mutations may have lower ejaculate volume, higher testosterone levels, higher oestradiol levels and higher androgen sensitivity index. However, the ranges for these variables are highly overlapping between men with and without AR gene mutations.56,57
AR defects in other conditions
A polymorphism involving the CAG triplet repeat expansion of the AR gene, coding for a polyglutamine (PolyQ) tract in the N-terminal transactivation domain of the AR protein, has been involved in certain endocrine and neurological disorders. In the endocrine disorders, the PolyQ size has been proposed as being associated with male infertility, hirsutism and cryptorchidism.58 The molecular mechanisms of these alterations are thought to involve a modulation of AR transcriptional competence, which inversely correlates with the PolyQ length.
Among neurological disorders, an expanded CAG repeat (greater than 35-40 contiguous glutamines) is linked to a rare inherited X-linked neurodegenerative disease, Spinal and Bulbar Muscular Atrophy (SBMA) or Kennedy’s disease.11,59 Kennedy’s disease is a chronic, progressive neuromuscular disorder characterized by proximal muscle weakness, atrophy and fasciculation. Affected males may show signs of androgen insensitivity, including gynaecomastia, reduced fertility and testicular atrophy. The principal pathological manifestation of SBMA is the loss of motor neurons in the spinal cord and brainstem.60
CAG repeat length (PolyQ) in the AR gene is also correlated with tumors of classical androgen target tissues, influencing the age at diagnosis, the total risk, the recurrence after surgery and the aggressive growth.61 In the prostate, where androgens exert a mitogenic effect, the cancer risk increases with decreasing PolyQ repeat length. In the breast, where androgens probably act as anti-mitogens, a higher risk and earlier onset of breast cancer has been reported in carriers of BRCA1 mutations, which also have long CAG repeats in the AR gene. Alterations in somatic cells leading to carcinogenesis appear to be frequent in endometrial and in colon cancer.61
ENDOCRINE FEATURES
The hormonal profiles of patients with CAIS and PAIS are identical. Serum testosterone (T) and luteinizing hormone (LH) are at or above the upper normal limit during the first 3 months of life, while prepubertal patients generally have T and LH concentrations in the normal range for their age.62,63 In CAIS, testosterone levels are elevated at the time of puberty. Elevated luteinizing hormone (LH) levels are found, indicating androgen resistance at the hypothalamic-pituitary level. The high levels of testosterone, a substrate for aromatase activity, result in substantial amounts of estrogens, which are responsible for very good breast development at puberty in CAIS individuals. Adults with in situ testes usually have increased levels of LH, normal (sometimes elevated) concentration of T and follicle-stimulated hormone (FSH) as compared to normal males, and estradiol at the upper normal limits.51,64 In PAIS, hCG testing is necessary to demonstrate normal T and DHT production so as to exclude defects in testosterone biosynthesis and 5alpha-reductase 2 deficiency. High levels of LH result from a reduced sensitivity of the hypothalamus and hypophysis to the negative feedback regulation of gonadotropin secretion by sex steroids, probably due to impaired androgen sensitivity. The increased LH secretion stimulates Leydig cell steroid production and results in increased production of testosterone and estradiol.65,66 In patients with AIS, Anti-M?llerian Hormone (AMH) concentration is normal as the secretion and function of sertoli cells is not impaired. However, rare sporadic cases of CAIS have been reported with M?llerian remnants and inappropriate synthesis or action of AMH.67
MOLECULAR DEFECTS IN AIS
AIS is associated with a wide variety of molecular defects that may or may not affect androgen binding. These include: (a) single point mutations resulting in amino acid substitution or premature stop codon; (b) nucleotide insertions or deletions most often leading to a frameshift and premature termination; (c) complete or partial gene deletions (>10 nucleotides); and (d) intronic mutations in either splice donor or acceptor sites, which affect the splicing of AR RNA. According to the AR mutation database (http://www.mcgill.ca/androgendb), there are more than 400 different mutations reported so far in the AR gene and the number is continuously increasing. The majority of these molecular defects are single base substitutions, while relatively few deletions or insertions have been detected. There is an unequal distribution of mutations along the length of the AR gene with hot spots located mainly in the LBD.68 The observed functional impact of a mutation in the AR gene depends on the exact locus across the gene sequence. Mutations in the NTD (exon 1 of the gene) do not occur frequently and the vast majority result in a stop codon or premature termination due to frameshifts caused by nucleotide insertions or deletions.13 The LBD, which is encoded by less than half of the AR gene, harbors the majority of mutations thus far identified. Mutations in exons 5 and 7 are the most frequent and in their vast majority are single base substitutions (Androgen Receptor Mutation Database, http://www.mcgill.ca/androgendb). A special group of interesting but rare mutations are the splice donor and splice acceptor site mutations in the AR gene as well as mutations outside the coding region.13,69
Most of the identified mutations of the AR gene are isolated, but they may also be recurrent. The majority of molecular defects are inherited (~70%), while about one third are de novo mutations, either germline or somatic.70 Germline mutations arise either from a single germ cell or a germ cell mosaicism of the mother, while somatic mutations indicate a molecular defect that was not inherited but occurred after the zygotic stage. In the latter case, the patient exhibits somatic mosaicism with both mutant and wild-type receptors expressed in different proportions. Somatic mutations in the AR gene have only recently been reported.71-75 Identification of a somatic mutation can be difficult as it may be present only in a small number of cells, but it is of great importance for the correct sex assignment at birth, since the presence of a functional wild-type AR can induce virilization at puberty.70
Beyond the well-defined molecular defects in the AR gene, cases have been described with a degree of androgen resistance, clinical phenotype of AIS but normal AR gene sequence. One of the variable pathophysiological mechanisms suggested in the literature for these cases is the expansion of the polyglutamine repeats within AR that may interact with other unknown factors and result in undermasculinization.32,76 In addition, the presence of a co-regulator protein defect may have a serious impact on the final phenotype, as pointed out by Adachi et al.77
PHENOTYPE-GENOTYPE CORRELATIONS OF AR MUTATIONS
The observed functional impact of a mutation in the AR gene on the phenotype depends on the exact locus of the defect across the gene sequence.52 Boehmer et al78 analysed the genotype-phenotype relationship in AIS and the possible causes of phenotypic variations in families with many affected individuals. Intrafamilial phenotypic variation was observed for mutations R846H and M771I. Patients with a functionally defective AR have some pubic hair, Tanner stage P2 and vestigial Wolffian duct derivatives despite absence of AR expression. Vaginal length was functional in most but not all CAIS patients. Boehmer et al concluded that, while phenotypic variation was absent in families with CAIS, distinct phenotypic variation was frequently observed in families with PAIS. According to literature reports, complete gene deletions, partial deletions, insertions, duplications as well as nonsense and frameshift mutations leading to a premature translation termination codon result almost exclusively in complete AIS. Truncated androgen receptors have been shown in vitro not to cause transactivation and/or ligand binding.79 Mutations that affect splicing are found in CAIS as well as in PAIS patients. The resulting phenotype depends on the residual function of the mutant AR and on the amount of functional or wild-type transcript produced by alternative splicing.80,81 Missense mutations, however, resulting in single amino acid substitutions, represent the most common structural defects associated with diverse phenotype.
Different factors have been suggested as influencing the expression of AR mutations. The traditional explanation is that the level of competence of co-regulatory proteins acts as a genetic “background” factor in determining the overall clinical outcome.52 Co-regulators are molecules that interact with nuclear receptors either to increase (co-activators) or to decrease (co-repressors) gene transcription in a ligand-dependent manner by forming a multiprotein complex which involves the basic transcription machinery.82,83 Furthermore, Holterhus et al84 have recently investigated a patient with PAIS phenotype and defective AR transcription and translation, despite the absence of a molecular abnormality in the entire AR gene. The authors attributed the reduced activity of the AR protein found in this case to the possible existence of a defective AR promoter caused by a mutation or by reduced cellular availability or dysfunction of an AR-promoter-interacting factor critical for the initiation of the AR transcription. Another probably important mechanism which may account for some variable expressivity is the presence of somatic mosaicism of mutant and wild-type alleles of AR, due to a de novo postzygotic somatic mutation.72,85 According to Hiort et al,71 the proportion of somatic mosaicism in a group of 30 families with AIS patients was relatively high (3 of the 8 patients had de novo mutations). Somatic mosaicism should always be considered in AIS individuals with unexpected normal virilization, which could be the result of the expression of the wild-type androgen receptor in some cell lines.86 Holterhus et al previously presented a case of a 46,XY newborn with ambiguous genitalia carrying a mosaic of Val866Met mutation with the wild-type AR gene. Despite the fact that this mutation has usually been associated with the complete AIS phenotype, the newborn exhibited signs of virilization due to the expressed wild-type receptor.85 Discrepancies of phenotype-genotype can also be caused by splice site mutations due to alternative splicing,81,87 or by differences in 5alpha-reductase 2 activity and thus in adequate DHT availability.78 The length of polyglutamine repeats in exon 1 is also a candidate factor leading to diverse phenotypes.88 In a recent report, Werner et al89 documented experimentally the contradictory effect of the combination of a short polyglycine (PolyG) repeat with the rare mutation of the hinge region A645D, resulting in seriously reduced AR activity when paired with a long polyglutamine (PolyQ) repeat and in almost wild-type AR activity when paired with a short PolyQ repeat.
DIAGNOSIS OF PATIENTS WITH AIS
The diagnosis of CAIS is usually based on clinical findings and laboratory evaluation. The diagnosis of PAIS and MAIS may additionally require a family history consistent with X-linked inheritance.52 To establish the diagnosis of AIS, a 46,XY karyotype is essential, as well as determination (baseline or after hCG stimulation) of testosterone, testosterone precursors and DHT levels, in order to exclude defects in testosterone biosynthesis or 5alpha-reductase 2 deficiency. The clinical phenotype can be classified according to Quigley et al in 7 grades (Table 1 ). Family history compatible with X-linkage can be helpful, especially in the partial forms of the disease.50 Manifesting carriers should also be identified through the detailed study of the family tree; these present with asymmetric distribution and sparse or delayed growth of pubic or axillary hair and account for about 10% of all carriers.90 In postpubertal patients, normal breast development and primary amenorrhea contribute to the clinical suspicion of AIS. Additional endocrine findings are discussed in the present text under “endocrine features”. Imaging studies in individuals with AIS and female phenotype show a short blind-end vagina, absence of uterus and other Müllerian remnants, as well as absence or underdevelopment of Wolffian duct derivatives, depending on the degree of the functional AR defect. The gonads will always be testes and can be found in any position, from the abdomen to the scrotum/labia majora, though they are most frequently found in the inguinal region.
The diagnosis of AIS is confirmed with the identification of a molecular defect in the AR gene. Sequence analysis of all 8 exons of the AR gene detects mutations in more than 95% of individuals with CAIS.90 Mutation detection rate for milder phenotypes is not known; in several studies, however, it ranges from 28%33 to 73%.49,50 Study of androgen-binding activity in genital skin fibroblasts could be helpful in the diagnosis of PAIS patients, increasing the likelihood of finding a mutation in the LBD when impaired.91
The variable expression within or among families is of great importance and is disclosed by taking a sophisticated family history. For instance, the presence of apparently isolated hypospadias or azoospermia, or maternally related females with delayed menarche, primary amenorrhea, delayed and reduced or absent sex hair, or even cliteromegaly with or without posterior labial fusion may be caused by androgen insensitivity. The equivalent would be true in the early differential diagnosis of phenotypic females with any of these presenting signs.52
Management of AIS patients
Sex assignment of children with ambiguous genitalia remains a difficult and complicated decision for the families and clinicians involved and is subject to controversy among professionals and self-help groups.92,93 These decisions must be based on the correct diagnosis, which will facilitate prediction of development during puberty and adulthood in affected individuals.94 Whether virilization will increase at puberty or following androgen therapy in a neonate with ambiguous genitalia is a crucial question. Kohler et al70 suggest a testosterone treatment trial in all patients with PAIS in order to evaluate the virilizing capacity of the newborn external genitalia before sex assignment. This is, nonetheless, a currently controversial topic among pediatric endocrinologists, because there is no evidence that a good response to exogenous testosterone in neonates will be followed by a similar response at puberty. However, in the case of an AR somatic mosaicism identified at birth, a testosterone treatment trial could be warranted because a certain type of the wild-type receptor is present and likely functionally active. In the same study, Kohler et al presented two PAIS patients with somatic mosaicism, who showed sufficient virilization at puberty due to the functional wild-type AR. In general, sex-of-rearing is assigned on the basis of genital phenotype and hormonal data, as reported above, as well as the clinical response to a testosterone treatment trial, the feasibility of reconstructive surgery and molecular studies of the AR gene. Depending on the phenotype, management of the patients is as follows:
CAIS: XY individuals with complete androgen insensitivity syndrome in whom the external genitalia are those of a normal female pose no dilemma of sex assignment. Affected individuals are raised as females, and in such cases the pediatric endocrinologist can handle the problem along with an experienced psychiatrist who will support both the family at diagnosis as well as the patient later in life.92
A common practice is to remove the testes after puberty when feminization of the affected individual is complete, since feminization occurs partly by testicular estrogen and partly by peripheral conversion of androgen to estrogen. The reason for the postpubertal gonadectomy is the risk of testicular malignancy, which seldom occurs before puberty. Goulis et al reported a case of bilateral testicular hamartomata in an 18-year-old individual with CAIS, who carried the R831X mutation of the AR gene.95 Prepubertal gonadectomy is indicated if inguinal testes are physically or esthetically uncomfortable and if inguinal herniorrhaphy is necessary. In this case, estrogen replacement therapy is necessary to initiate puberty, maintain feminization and avoid osteoporosis. Vaginal length may be short and require dilatation in an effort to avoid dyspareunia. Wisniewski et al96 assessed by questionnaire and medical examination the physical and psychosexual status of 14 women with documented CAIS. Secondary sexual development of these women was found satisfactory, as judged by both participants and physicians. Most women were satisfied with their psychosexual development and sexual function. All of the women who participated were satisfied with having being raised as females and none desired a gender reassignment. In general, the medical, surgical and psychosexual outcomes for women with CAIS were satisfactory; nonetheless, specific ways for improving long-term treatment of this population were identified.
PAIS: In PAIS with predominantly female genitalia, the issues are similar to those discussed under CAIS, with the exception that prepubertal gonadectomy is preferred to avoid the emotional discomfort of increasing clitoromegaly at the time of puberty. In PAIS with ambiguous genitalia or predominantly male genitalia, sex assignment is a complex process that requires timely assessment by a multidisciplinary team in consultation with the family and should be resolved as early as feasible. Aside from purely anatomical and surgical considerations, the choice of a male sex-of-rearing demands a therapeutic trial with testosterone in an effort to predict potential androgen responsiveness at puberty. Furthermore, appreciable phallic growth in response to administered androgen facilitates reconstructive surgery.97
MAIS: In infertile subjects with MAIS, promotion of spermatogenesis would be the main goal, and sperm retrieval would not be necessary in the context of an intact sperm-delivery system. Apart from one successful experience with mesterolone (1a-methylandrostan-17β-ol-3-one) in promoting spermatogenesis and fertility twice in a man with MAIS,98 experience with long-term natural androgen pharmacotherapy is meager and its value unclear.44,99-101
GENETIC COUNSELING
Most cases (70%) of AR mutations are inherited and transmitted in an X-linked manner. In this situation, there is a likelihood of 50% for an XY offspring of being affected and for an XX offspring of being a healthy carrier. De novo mutations represent 30% of AR mutations and the risk of transmission can be considered as being very low, since there is to date no report of transmission to a second child. A rare exception to this (only two reported instances) is the case of germline de novo mutations in the mother. In this circumstance, the presence of germ cell mosaicism can be assumed and the risk of transmission is regarded as high.70,75 However, the possibility of germline mosaicism cannot be excluded in any case of de novo mutation of the AR gene and one should be cautious in the genetic counseling of these families.
REFERENCES
1. Morris JM, 1953 The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol 65: 1192-1211.
2. Lyon MF, Hawkes SG, 1970 X-linked gene for testicular feminization in mouse. Nature 227: 1217-1219.
3. Ohno S, Lyon MF, 1970 X-linked testicular feminization in the mouse as a non-inducible regulatory mutation of the Jacob-Monod type. Clin Genet 1: 121-127.
4. Migeon BR, Brown TR, Axelman J, Migeon CJ, 1981 Studies of the locus for androgen receptor: Localization on the Human X chromosome and evidence for homology with the Tfm locus in the mouse. Proc Natl Acad Sci USA 78: 6339-6343.
5. Brown CJ, Goss SJ, Lubahn DB, et al, 1989 Androgen receptor locus on the human X chromosome: regional localization to Xq11-12 and description of a DNA polymorphism. Am J Hum Genet 44: 264-269.
6. Brown TR, Lubahn DB, Wilson EM, Joseph DR, French FS, Migeon CJ, 1988 Deletion of the steroid-binding domain of the human androgen receptor gene in one family with complete androgen insensitivity syndrome: evidence for further genetic heterogeneity in this syndrome. Proc Natl Acad Sci USA 85: 8152-8155.
7. Lubahn DB, Brown TR, Simantal JA, et al, 1989 Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity. Proc Natl Acad Sci USA 86: 9534-9538.
8. Kuiper GCJM, Faber PW, van Rooij HJC, et al, 1989 Structural organization of the human androgen receptor gene. J Mol Endocrinol 2: R1-R4.
9. Brinkmann AO, Faber PW, van Rooij HJC, et al, 1989 The human androgen receptor: domain structure, genomic organization, and regulation of expression. J Steroid Biochem 34: 307-310.
10. Marcelli M, Tilley WD, Wilson CM, Griffin JE, Wilson JD, McPhaul MJ, 1990 Definition of the human androgen receptor gene structure permits the identification of mutations that cause androgen resistance: premature termination of the receptor protein at amino acid residue 588 causes complete androgen resistance. Mol Endocrinol 4: 1105-1116.
11. La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH, 1991 Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352: 77-79.
12. Hiort O, Holterhus PM, 2003 Androgen insensitivity and male infertility. Int J Androl 26: 16-20.
13. Brinkmann AO, 2001 Molecular basis of androgen insensitivity. Mol Cell Endocrinol 179: 105-109.
14. McPhaul MJ, Griffin JE, 1999 Male pseudohermaphroditism caused by mutations of the human androgen receptor. J Clin Endocrinol Metab 84: 3435-3441.
15. Evans RM, 1988 The steroid and thyroid hormone receptor superfamily. Science 240: 889-895.
16. Mangelsdorf DJ, Thummel C, Beato M, 1995 The nuclear receptor superfamily: the second decade. Cell 83: 835-839.
17. Warne GL, Kanumakala SH, 2002 Molecular endocrinology of sex differentiation. Semin Reprod Med 20: 169-180.
18. Sultan C, Paris F, Terouanne B, et al, 2001 Disorders linked to insufficient androgen action in male children. Hum Reprod Update 7: 314-322.
19. Jester G, van der Korput HAGM, van Vroonhooven C, van der Kwast TH, Trapman J, Brinkmann AO, 1991 Domains of the human androgen receptor involved in steroid binding, transcriptional activation and subcellular localization. Mol Endocrinol 5: 1396-1404.
20. Simental JA, Sar M, Lane MV, French FS, Wilson EM, 1991 Transcription activation and nuclear targeting signals of the human androgen receptor. J Biol Chem 266: 510-518.
21. Rundlett SE, Wu X-P, Miesfeld RL, 1990 Functional characterizations of the androgen receptor confirm that the molecular basis of androgen action is transcriptional regulation. Mol Endocrinol 4: 708-714.
22. Palvimo JJ, Kallio PJ, Ikonen T, Mehto M, Janne OA, 1993 Dominant negative regulation of trans-activation by the rat androgen receptor: roles of the N-terminal domain and heterodimer formation. Mol Endocrinol 7: 1399-1407.
23. Freedman LP, 1992 Anatomy of the steroid receptor zinc finger region. Endocr Rev 13: 129-145.
24. Berg J, 1989 DNA-binding specificity of steroid receptors. Cell 57: 1065-1068.
25. Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS, 1995 Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev 16: 271-321.
26. Zhou Z-X, Sar M, Simental JA, Lane MV, Wilson EM, 1994 A ligand-dependent bipartite nuclear targeting signal in the human andogen receptor. J Biol Chem 269: 13115-13123.
27. Matias PM, Donner P, Coelho R, et al, 2000 Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem 275: 26164-26271.
28. Georget V, Lobaccaro JM, Terouanne B, Mangeat P, Nicolas JC, Sultan C, 1997 Trafficking of the androgen receptor in living cells with fused green fluorescent protein-androgen receptor. Mol Cell Endocrinol 129: 17-29.
29. Meehan KL, Sadar MD, 2003 Androgens and androgen receptor in prostate and ovarian malignancies. Front Biosci 8: d780-800.
30. Sultan C, Savage MO 1998. In Grossman A (ed), Clinical Endocrinology, Blackwell Science, Oxford; pp, 795-809.
31. Savage MO, Chaussain JL, Evain D, Roger M, Canlorbe P, Job JC, 1978 Endocrine studies in male pseudohermaphroditism in childhood and adolescence. Clin Endocrinol (Oxf) 8: 219-231.
32. Ahmed SF, Hughest IA, 2002 The genetics of male undermasculinization. Clin Endocrinol 56: 1-18.
33. Ahmed SF, Cheng A, Dovey L, et al, 2000 Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab 85: 658-665.
34. Ulloa-Aguirre A, Carranza-Lira S, Mendez JP, Angeles A, Chavez B, Perez-Palacios G, 1990 Incomplete regression of Mullerian ducts in the androgen insensitivity syndrome. Fertil Sertil 53: 1024-1028.
35. Dodge ST, Finkelstone MS, Miyazawa K, 1985 Testicular feminization with incomplete Mullerian regression. Fertil Sertil 43: 937-938.
36. Swanson ML, Coronel EH, 1993 Complete androgen insensitivity with persistent Mullerian structures. A case report. J Reprod Med 38: 565-568.
37. Rutgers JL, Scully RE, 1991 The androgen insensitivity syndrome (testicular feminization): a clinicopathological study of 43 cases. Int J Gynecol Pathol 10: 126-144.
38. Galani A, Sofocleous C, Karahaliou F, Papathanasiou A, Kitsiou-Tzeli S, Kalpini-Mavrou A, 2008 Sex-reversed phenotype in association with two novel mutations c.2494delA and c.T3004C in the ligand-binding domain of the androgen receptor gene. Fertil Steril [Epub ahead of print]
39. Bale PM, Howard NJ, Wright GE, 1992 Male pseudohermaphroditism in XY children with female phenotype. Pediatr Pathol 12: 29-49.
40. Morris JM, Mahesh VB, 1963 Further observations on the syndrome “testicular feminization”. Am J Obstet Gynecol 87: 731-748.
41. Wilkins L 1950 Heterosexual development. In: The diagnosis and treatment of endocrine disorders in childhood and adolescence. Charles C Thomas, Springfied, IL; pp, 256-279.
42. Papadimitriou DT, Linglart A, Morel Y, Chaussain JL, 2006 Puberty in subjects with complete androgen insensitivity syndrome. Horm Res 65: 126-131.
43. Stephens JD, 1984 Prenatal diagnosis of testicular feminization. Lancet 2: 1038.
44. Hiort O, Huang Q, Sinnecker GHG, et al, 1993 Single strand conformational polymorphism analysis of androgen receptor gene mutations in patients with androgen insensitivity syndromes: application for diagnosis, genetic counselling and therapy. J Clin Endocrinol Metab 77: 262-266.
45. Grumbach MM, Conte FA 1991 Disorders of sex differentiation. In: Wilson JD, Foster DW(eds) Williams textbook of endocrinology, Saunders, Philadelphia; pp, 853-951.
46. Aiman J, Griffin JE, Gazak JM, Wilson JD, MacDonald PC, 1979 Androgen insensitivity as a cause of infertility in otherwise normal men. N eng J Med 300: 223-227.
47. Aiman J, Griffin JE, 1982 The frequency of androgen receptor deficiency in infertile men. J Clin Endocrinol Metab 54: 725-732.
48. Morrow AF, Gyorky S, Warne GL, et al, 1987 Variable androgen receptor levels in infertile men. J Endocrinol Metab 64: 1115-1121.
49. Ferlin A, Vinanzi C, Garolla A, et al, 2006 Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations. Clin Endocrinol (Oxf) 65: 606-610.
50. Melo KF, Mendonca BB, Billerbeck AE, et al, 2003 Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort: five novel mutations in the androgen receptor gene. J Clin Endocrinol Metab 88: 3241-3250.
51. Balducci R, Ghirri P, Brown TR, et al, 1996 A clinician looks at androgen resistance. Steroids 61: 205-211.
52. Gottlieb B, Pinsky L, Beitel LK, Trifiro M, 1999 Androgen insensitivity. Am J Med Genet 89: 210-217.
53. Migeon CJ, Brown TR, Lanes R, Palacios A, Amrhein JA, Schoen EJ, 1984 A clinical syndrome of mild androgen insensitivity. J Clin Endocrinol Metab 59: 672-678.
54. Cundy TF, Rees M, Evans BAJ, Hughes IA, Butler J, Wheeler MJ, 1986 Mild androgen insensitivity presenting with sexual dysfunction. Fertil Steril 46: 721-723.
55. Pinsky L, Kaufman K, Killinger DW, Burko B, Shatz D, Volpe R, 1984 Human minimal androgen insensitivity with normal dihydrosterone-binding capacity in cultured genital skin fibroblasts: evidence for an androgen-selective qualitative abnormality of the receptor. Am J Hum Genet 36: 965-978.
56. Zuccarello D, Ferlin A, Vinanzi C, et al, 2008 Detailed functional studies on androgen receptor mild mutations demonstrate their association with male infertility. Clin Endocrinol (Oxf) 68: 580-588.
57. Ferlin A, Vinanzi C, Garolla A, et al, 2006 Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations. Clin Endocrinol (Oxf) 65: 606-610.
58. Palazzolo I, Gliozzi A, Rusmini P, et al, 2008 The role of the polyglutamine tract in androgen receptor. J Steroid Biochem Mol Biol 108: 245-253.
59. Vottero A, Capelletti M, Giuliodori S, et al, 2006 Decreased androgen receptor gene methylation in premature pubarche: a novel pathogenetic mechanism? J Clin Endocrinol Metab 91: 968-972.
60. Li M, Sobue G, Doyu M, Mukai E, Hashizume Y, Mitsuma T, 1995 Primary sensory neurons in X-linked recessive bulbospinal neuropathy: histopathology and androgen receptor gene expression. Muscle Nerve 18: 301-308.
61. Ferro P, Catalano MG, Dell’Eva R, Fortunati N, Pfeffer U, 2002 The androgen receptor CAG repeat: a modifier of carcinogenesis? Mol Cell Endocrinol 193: 109-120.
62. Balducci R, Adamo M, Mangiantini A, Municchi G, Toscano V, 1986 Testicular responsiveness to a single hCG dose in patients with testicular femiization. Horm Metabol Res 21: 449-452.
63. Campo S, Stivel M, Nicolau G, Monteagudo C, Rivarola M, 1979 Testicular function in postpubertal male pseudohermaphroditism. Clin Endocrinol (Oxf) 11: 481-490.
64. Imperato-McGinley J, Peterson RE, Gautier T, et al, 1982 Hormonal evaluation of a large kindred with complete androgen insensitivity syndrome: evidence for secondary 5a-reductase deficiency. J Clin Endocrinol Metabol 54: 931-941.
65. Griffin JE, Wilson JD 1989 The androgen resistance syndromes: 5a-reductase deficiency, testicular feminization, and related disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic basis of inherited disease, McGraw-Hill, New York; pp, 1919-1944.
66. Boyar RM, Moore RJ, Ronser W, et al, 1978 Studies of gonadotrophin-gonadal dynamics in patients with androgen insensitivity. J Clin Endocrinol Metab 47: 1116-1122.
67. Van YH, Li JL, Huang SF, Luo CC, Hwang CS, Lo FS, 2003 Novel point mutations in complete androgen insensitivity syndrome with incomplete m?llerian regression: two Taiwanese patients. Eur J Pediatr 162: 781-784.
68. Gottlieb B, Beitel LK, Wu JH, Trifiro, 2004 The androgen receptor gene mutations database (ARDB): 2004 update. Hum Mutat 23: 527-533.
69. Sammarco I, Grimaldi P, Rossi P, et al, 2000 Novel point mutation in the splice donor site of exon-intron junction 6 of the androgen receptor gene in a patient with partial androgen insensitivity syndrome. J Clin Endocrinol Metab 85: 3256-3261.
70. Kohler B, Lumbroso S, Leger J, et al, 2005 J Clin Endocrinol Metab 90: 106-111.
71. Hiort O, Sinnecker GH, Holterhus PM, Nitsche EM, Kruse K, 1998 Inherited and de novo androgen receptor gene mutations: investigation of single-case families. J Pediatr 132: 939-943.
72. Holterhus PM, Bruggenwirth HT, Hiort O, et al, 1997 Mosaicism due to a somatic mutation of the androgen receptor gene determines phenotype in androgen insensitivity syndrome. J Clin Endocrinol Metab 82: 3584-3589.
73. Gottlieb B, Beitel LK, Trifiro MA, 2001 Somatic mosaicism and variable expressivity. Trends Genet 17: 79-82.
74. Holterhus PM, Wiebel J, Sinnecker GH, et al, 1999 Clinical and molecular spectrum of somatic mosaicism in androgen insensitivity syndrome. Pediatr Res 46: 684-690.
75. Boehmer AL, Brinkmann AO, Niermeijer MF, Bakker L, Halley DJ, Drop SL, 1997 Germ-line and somatic mosaicism in the androgen insensitivity syndrome: implications for genetic counselling. Am J Hum Genet 60: 1003-1006.
76. Ogata T, Muroya K, Ishii T, Suzuki Y, Nakada T, Sasagawa I, 2001 Undermasculinized genitalia in a boy with an abnormally expanded CAG repeat length in the androgen receptor gene. Clin Endocrinol 54 : 835-838.
77. Adachi M, Takayanagi R, Tomura A, et al, 2000 Androgen insensitivity syndrome as a possible coactivator disease. N Eng J Med 343: 856-862.
78. Boehmer AL, Brinkmann AO, Nijman RM, et al, 2001 Phenotypic variation in a family with partial androgen insensitivity syndrome explained by differences in 5alpha dihydrotestosterone availability. J Clin Endocrinol Metab 86: 1240-1246.
79. Jenster J, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO, 1991 Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol 5: 1396-1404.
80. Ris-Stalpers C, Turberg A, Verleun-Mooyman MC, 1993 Expression of an aberrantly spliced androgen receptor mRNA in a family with complete androgen insensitivity. Ann N Y Acad Sci 684: 239-242.
81. Ris-Stalpers C, Verleun-Mooyman MC, de Blaeij TJ, Degenhart HJ, Trapman J, Brinkmann AO, 1994 Differential splicing of human androgen receptor pre-mRNA in X-linked Reifenstein syndrome, because of a deletion involving a putative branch site. Am J Hum Genet 54: 609-617.
82. McKenna NJ, Xu J, Nawaz Z, Tsai SY, Tsai MJ, O’Malley BW, 1999 Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J Steroid Biochem Mol Biol 69: 3-12.
83. Robyr D, Wolffe AP, Wahli W, 2000 Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol Endocrinol 14: 329-347.
84. Holterhus PM, Werner R, Hoppe U, et al, 2005 Molecular features and clinical phenotypes in androgen insensitivity syndrome in the absence and presence of androgen receptor gene mutations. J Mol Med 83: 1005-1013.
85. Holterhus PM, Sinnecker GH, Wollmann HA, et al, 1999 Expression of two functionally different androgen receptors in a patient with androgen insensitivity. Eur J Pediatr 158: 702-706.
86. Holterhus PM, Wiebel J, Sinnecker GH, et al, 1999 Clinical and molecular spectrum of somatic mosaicism in androgen insensitivity syndrome. Pediatr Res 46: 684-690.
87. Sammarco I, Grimaldi P, Rossi P, et al, 2000 Novel point mutation in the splice donor site of exon-intron junction 6 of the androgen receptor gene in a patient with partial androgen insensitivity syndrome. J Clin Endocrinol Metab 85: 3256-3261.
88. Knoke I, Allera A, Wieacker P, 1999 Significance of CAG repeat length in the androgen receptor gene (AR) for the transactivation function of an M7801 mutant AR. Hum Genet 104: 257-261.
89. Werner R, Holterhus PM, Binder G, et al, 2006 J Clin Endocrinol Metab 91: 3515-3120.
90. Gottlieb B, Beitel L, Trifiro MA, 2007 Androgen insensitivity syndrome. www.geneclinics.org
91. Weidemann W, Linck B, Haupt H, et al, 1996 Clinical and biochemical investigations and molecular analysis of subjects with mutations in the androgen receptor gene. Clin Endocrinol (Oxf) 45: 733-739.
92. Dacou-Voutetakis C, 2007 A multidisciplinary approach to the management of children with complex genital anomalies. Nat Clin Pract Endocrinol Metab 3: 668-669.
93. Thyen U, Richter-Appelt H, Wiesemann C, Holterhus PM, Hiort O, 2005 Deciding on gender in children with intersex conditions: considerations and controversies. Treat Endocrinol 4: 1-8.
94. Hiort O, Reinecke S, Thyen U, et al, 2003 Puberty in disorders of somatosexual differentiation. J Pediatr Endocrinol Metab 16: Supp 2: 297-306.
95. Goulis DG, Iliadou PK, Papanicolaou A, et al, 2006 R831X mutation of the androgen receptor gene in an adolescent with Complete Androgen Insensitivity Syndrome and bilateral testicular hamartomata. Hormones (Athens) 5: 200-204.
96. Wisniewski AB, Migeon CJ, Meyer-Bahlburg HF, et al, 2000 Complete androgen insensitivity syndrome: long-term medical, surgical, and psychosexual outcome. J Clin Endocrinol Metab Aug 85: 2664-2669.
97. Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES Consensus Group, ESPE Consensus Group, 2006 Consensus statement on management of intersex disorders. Arch Dis Child 91: 554-563.
98. Yong EL, Ng SC, Roy AC, Yun G, Ratnam SS, 1994 Pregnacy after hormonal correction of severe spermatogenic defect due to mutation in androgen receptor gene. Lancet 344: 826-827.
99. Juckier L, Kaufman M, Prinsky L, Peterson RE, 1984 Partial androgen resistance associated with secondary 5a-reductase deficiency: identification of a novel qualitative androgen receptor defect and clinical implications. J Endocrinol Metab 59: 679-688.
100. Price P, Wess JAH, Griffin JE, et al, 1984 High dose androgen therapy in male pseudohermaphroditism due to 5a-reductase deficiency and disorders of the androgen receptor. J Clin Invest 74: 1496-1508.
101. Mc Phaul MJ, Marcelli M, Tilley WD, Griffin JE, Isidro-Gutierrez RF, Wilson JD, 1991 Molecular basis of androgen resistance in a family with a qualitative abnormality of the androgen receptor and responsive to high-dose androgen therapy. J Clin Invest 87: 1413-1421.
Address for correspondence:
Galani Angeliki, “Choremis” Research Laboratory, Department of Medical Genetics,
Athens University School of Medicine, “Agia Sophia” Children’s Hospital,
Thivon and Levadias, 115 27 Athens, Greece, Tel: +30 210 7467462,
Fax: +30 210 7795553, e-mail: agalani@med.uoa.gr
Received 22-07-07, Revised 15-03-08, Accepted 10-04-08