1Unit of Reproductive Endocrinology, First Department of Obstetrics and Gynecology, Papageorgiou General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece, 2Laboratory of Genetics, Second Department of Obstetrics and Gynecology, University of Ioannina, Ioannina, Greece
An 18-year old, phenotypically female individual was examined for primary amenorrhea. Three months before her referral, the patient underwent surgery and a pelvic mass was removed. The physical examination revealed normal female external genitalia, normal breast development, sparse pubic hair and absence of axillary hair. The gynecological examination revealed a short blind vagina pouch and absence of cervix and uterus. Serum testosterone and dihydrotestosterone levels were very high. Karyotype was that of a normal male (46,XY). The transabdominal ultrasound, computed tomography (CT) and Magnetic resonance imaging (MRI) showed absence of uterus and fallopian tubes and revealed testis-like gonads located at the internal opening of the inguinal canal bilaterally. Bilateral gonadectomy was subsequently performed. The pathology report was that of .hamartomatous testes. and associated paratesticular leiomyoma. The clinical, laboratory, imaging, genetic and histological findings confirmed the diagnosis of complete androgen insensitivity syndrome. DNA analysis revealed a R831X mutation in exon 7 of the androgen receptor gene. A Sertoli-cell dynamic test showed elevated basal serum inhibin-B and anti-Müllerian hormone levels without further rise following FSH stimulation. The patient was started on hormone replacement therapy with conjugated estrogens. Complete androgen insensitivity syndrome must be considered in any case of primary amenorrhea. Gonadectomy must be planned to eliminate the risk of gonadal malignancy.
Androgen insensitivity, Androgen receptor, Inhibin-B, Testicular hamartoma, Testicular leiomyoma
CASE REPORT
An 18-year old, phenotypically female individual was referred to the Unit of Reproductive Endocrinology due to primary amenorrhea. A year before her referral, a progestogen challenge test and a combined administration of estrogen and progestogen failed to induce menstrual bleeding. Three months before her referral, a transabdominal ultrasound scan revealed a pelvic mass. The patient underwent surgical removal of the mass (operation1), which was described as an .ovarian cystadenofibroma. in the pathology report.
Basal hormonal levels before the operation were: follicle-stimulating hormo
evels of prolactin, thyroid-stimulating hormone, free triidothyronine and free thyroxine were within the normal range. Karyotype was that of a normal male (46,XY). At this time she was referred to our Unit for further investigation.
The patient was born after an uncomplicated pregnancy with normal female external genitalia. Except for the primary amenorrhea, there were no other major events in her personal medical history. The physical examination revealed normal female external genitalia, normal breast development (Tanner stage IV . V), sparse pubic hair and absence of axillary hair. The gynecological examination revealed a short blind vagina pouch and absence of cervix and uterus. No hernias or palpable masses were found in either of the inguinal areas. The serum dihydrotestosterone (DHT) level was 1.9 nmol/ L (normal range: 0.17-0.76 nmol/L). A Sertoli-cell test (measurement of serum inhibin-B and anti-Mullerian hormone levels before and 24h and 48h after administration of 300 IU rhFSH subcutaneously) showed inhibin-B levels of 238 pg/ml at baseline, 219 pg/ml at 24h and 157 pg/ml at 48 h (normal range of basal values, as estimated in our Unit on a group of 20 normal men: 80-180 pg/ml at baseline). Serum anti-Müllerian hormone (AMH) levels were 10.27 ng/ml at baseline, 11.53 ng/ml at 24h and 11.95 ng/ml at 48h (normal range of basal values, as estimated in our Unit on a group of 10 normal men: 5.10-13.30 ng/ml at baseline). The transabdominal ultrasound, CT and MRI scans showed absence of uterus and fallopian tubes and revealed testis-like gonads (solid tissue with no apparent follicles) located at the internal opening of the inguinal canals bilaterally.
At this stage the provisional clinical diagnosis of Complete Androgen Insensitivity Syndrome (CAIS; former nomenclature: testicular feminization) was made based on the female phenotype, high levels of T and DHT, absence of Müllerian duct derivatives and 46,XY karyotype. The patient was referred for bilateral gonadectomy which was carried out by laparotomy. The pathology report of operation-1 was re-evaluated following the histopathologic examination of the removed gonads. The new combined pathology report was that of "hamartomatous testicles". Testicular parenchyma was replaced by hamartomatous nodules, which were separated by fibrous tissue (Figure 1). Microscopically the gonads consisted of immature seminiferous tubules lined by immature Sertoli cells, spindle-cell stroma resembling ovarian stroma and prominent Leydig cells. The immature tubules contained rare spermatogonia.
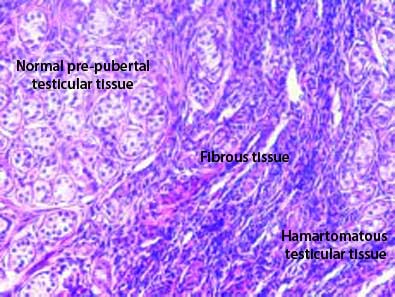
Figure 1. "Hamartomatous testicles" consist of all components of normal testicular parenchyma (seminiferous tubules and interstitial tissue) with disrupted architecture. In the upper left part of the picture the seminiferous tubules are normal but prepubertal; in the middle there is excess fibrous tissue and in the lower right part the architecture of the seminiferous tubules is disrupted.
Sertoli and Leydig cells were immunoreactive for inhibin (Figure 2) and calretinin (Figure 3), respectively. The investigation for atypical germ cells using the immunohistochemical staining of placental alkaline phosphatase was negative. A serous cyst and a tumor with the microscopic characteristics of a leiomyoma were adjacent to the left gonad. It became evident that the majority of the testicular tissue of the left gonad had been removed during operation-1.
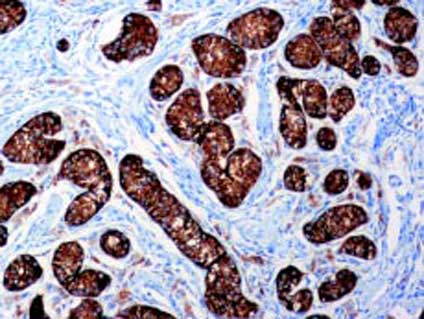
Figure 2. Inhibin immunohistochemical staining for Sertoli cells.Only Sertoli cells are positive for inhibin.
After operation-2, the patient.s serum FSH was 36.8 IU/L, LH 58.7 IU/L, O <0.7 nmol/L, estradiol (E2) 114 pmol/L, basal inhibin-B 14 pg/ml and AMH 1.41 ng/ml.
DNA was extracted from peripheral blood lymphocytes according to the standard salt extraction procedure. All eight exons of the Androgen Receptor (AR) gene were amplified using specific oligonucleotide primers. Polymerase chain reaction - single strand conformation polymorphism (PCR SSCP) analysis was used as the screening method for genetic variants. The unusual SSCP pattern found in exon 7 of the AR gene led to sequencing by ABI 3700 DNA Automated Sequencer. On sequencing, the AR gene was found to have a single nucleotide substitution, a C to T transition in codon 831 (exon 7 of the AR gene), resulting in a missing arginine and a stop codon (R831X).
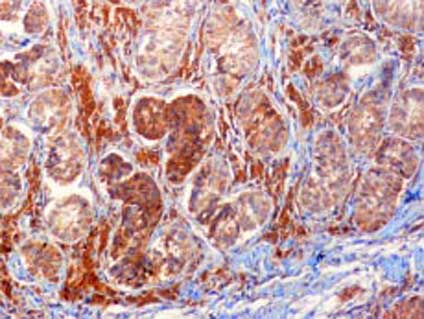
Figure 3. Calretinin immunohistochemical staining for Leydigcells. Leydig cells are strongly positive, whereas the Sertoli cells in the seminiferous tubules are weakly positive for calretinin.
The genetic and histological findings confirmed the diagnosis of CAIS. The patient was started on Hormone Replacement Therapy with conjugated estrogen (Premarin®) 0.625 mg daily.
DISCUSSION
The androgen insensitivity syndrome (AIS) is a rare (approximately 1:20,000 male births),1 X-linked disorder characterized by 46,XY karyotype, presence of normal testes and normal androgen production and metabolism. It is caused by a defect in the androgen receptor (AR) gene, which results in complete (CAIS) or partial (PAIS) androgen insensitivity with variable phenotypic expression. While individuals with CAIS have female external genitalia, PAIS cases have variable genitalia ambiguity. In the case of PAIS, if the gonads have not descended into the scrotum, the patients must undergo gonadectomy to eliminate the risk of gonadal malignancy.2
The AR gene is located on chromosomal locus Xq11-q12. More than 300 mutations have been described in patients with AIS, which include complete and partial gene deletions, point mutations and small insertions/deletions.3 It is, however, unclear whether there is a relationship between the site and type of mutation and the abnormality in androgen binding.4 Prenatal diagnosis as well as decisions about sex of rearing are hindered because of the clinical heterogeneity of phenotype for a specific mutation.
In our patient a previously reported mutation (R831X) of the androgen receptor gene was identified. SSCP analysis permits detection of mutations including single-base substitutions4 and is followed by direct DNA sequencing in the event the products show abnormal conformation. The AR gene is composed of eight exons, of which exon 1 encodes the N-terminal transcriptional activation domain, exons 2 and 3 encode the DNA-binding domain and exons 4-8 encode the C-terminal ligand-binding domain. In our patient a point mutation (R831X) in the ligand-binding domain resulted in a stop codon and premature termination. In codon 831 mutations R831X, R831Q and R831L have been found in CAIS (Androgen Receptor Gene Mutations Database, website: http://www.mcgill.ca/androgendb/, accessed on 18/3/2006). It is of interest to mention that, as in our patient, R831X mutation has also been described in another pubertal patient with early development of testicular hamartoma.5
Individuals with CAIS present at various stages of life, the timing of presentation having changed over the past 50 years as the index of suspicion for the diagnosis has increased. A very common presentation is the development of inguinal hernia, often bilateral, during infancy.4 A proportion of individuals, undiagnosed throughout childhood, present after puberty with primary amenorrhea, as was the case in this report. Karyotypic male individuals with CAIS have unambiguously female external genitalia, sparse or absent axillary and pubic hair, a blindly-ended vagina (which may be shortened or, less often, of normal depth), no uterus and normal breast development at puberty. The testes may be located at any site along the normal course of testicular descent: abdominal, inguinal or in the labia majora. In some cases remnants of the Mullerian or Wolffian ducts or their derivatives may be found.6
Classically, although not universally, postpubertal individuals with CAIS have increased serum concentrations of LH, normal or elevated serum concentrations of FSH, T similar to those of normal males and upper limit of normal or elevated blood or urinary estrogens.7 The LH-dependent stimulation of Leydig cells increases both the testicular androgens and estrogens and simultaneously the increased testosterone is aromatized to estradiol.
Consequently, the increased estrogens induce breast growth. In addition, our patient had received a combination of estrogen and progestogen for four months in the past. Hence, as expected, the breast development was normal. Gonadectomy results in a marked increase in LH and FSH levels indicating that some testis-mediated negative feedback suppression of hypothalamic GnRH secretion is present in individuals with AIS. In our patient the baseline FSH levels were low before operation-1, probably on account of the combined therapy with estradiol valerate and norgestrel. A significant increase in LH and FSH levels was observed after operation-1, during which a large part of the left gonad was removed. Additional increase was observed after complete bilateral gonadectomy (operation-2).
To the best of our knowledge, this is the first report of a Sertoli-cell dynamic test8 in a case of CAIS. As expected, the basal levels of inhibin-B exceed that of normal men in a mechanism similar to gonadal steroid levels.9 These results indicate that Sertoli cells are probably fully functional despite the hamartomatous nature of the testicles and that they are at maximal stimulation, as the administration of rhF-SH did not result in further increase of serum inhibin-B. According to some researchers the chronic gonadotropin stimulation may play a role in the induction of carcinogenesis in CAIS.10 Serum AMH levels have also been found elevated during puberty in individuals with CAIS, probably due to lack of T action, which is a major AMH inhibitor.11
Testicular tumors of germ cell and non-germ cell precursors occur with increased frequency in individuals with AIS, although series are small and it is unclear whether the incidence is any greater than that seen in simple cryptorchidism.2 Germ cell neoplasia is usually low grade, such as intratubular germ cell neoplasia (former nomenclature: carcinoma in situ), but seminomas (also called germinomas or dysgerminomas) have occasionally been reported, especially in postpubertal patients.12 The youngest such patient was a 14-year old girl.13 These individuals tend to be more than 30 years old and the risk of malignant germ cell tumors increases with age, ranging from about 3% at 20 years to 30% at 50 years.12 Tumors of non-germ cell origin also tend to be low grade, the most frequent being adenomas of Sertoli or Leydig cells. Sertoli-cell adenomas are common, occurring in 23% of the cases.14 Although Leydig-cell adenomas are less common, Leydig-cell hyperplasia is well recognized, as chronic gonadotropin stimulation may play a role in induction of Leydig-cell hyperplasia and subsequent neoplasia.10 Benign lesions, such as hamartomatous nodules, consisting of all components of normal testicular parenchyma but with a disrupted architecture, can also be found, as occurred in our patient.14 Inhibin and calretinin15 were used for immunohistochemical staining of Sertoli and Leydig cells, respectively, suggesting that they constitute valuable markers of normal and neoplastic testicular tissue. Finally, the co-existence of testicular hamartoma with paratesticular leiomyoma, as in our case, seems to be an extremely rare finding.16
In conclusion, in an 18-year old phenotypically female individual with primary amenorrhea, CAIS caused by R831X mutation of the AR gene was detected. In addition to displaying the well-described features of CAIS, basal serum inhibin-B levels were increased without further rise past FSH stimulation. Bilateral testicular hamartomas and a paratesticular leiomyoma were present. CAIS must be considered in any case of primary amenorrhea. Gonadectomy must be carried out to eliminate the risk of malignancy.
ACKNOWLEDGMENTS
We would like to thank Dr. John Tsitourides, Dr. Paris Polychronou and Ms. Vassiliki Pavlidou for their invaluable help during the clinical management of the case.
REFERENCES
1. Bangsboll S, Qvist I, Lebech PE, Lewinsky M, 1992 Testicular feminization syndrome and associated gonadal tumors in Denmark. Acta Obstet Gynecol Scand
71: 63-66.
2. Quigley CA, De Bellis A, Marschke KB, El-Awady MK, Wilson EM, French FS, 1995 Androgen receptor defects: historical, clinical and molecular perspectives. Endocr Rev 16: 271.321.
3. Gottlieb B, Levaslaiho H, Beitel LK, Lumbroso R, Pin-sky L, Trifiro M, 1998 The androgen receptor gene mutations database. Nucleic Acids Res 26: 234.238.
4. Ahmed SF, Cheng A, Dovey L, et al, 2000 Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as Androgen Insensitivity Syndrome. J Clin Endocrinol Metab 85: 658.665.
5. Chen CP, Chern SR, Chen BF, Wang W, Hwu YM, 2000 Hamartoma in a pubertal patient with complete androgen insensitivity syndrome and R(831)X mutation of the androgen receptor gene. Fertil Steril 74: 182.183.
6. Hannema SE, Scott IS, Hodapp J, et al, 2004 Residual activity of mutant androgen receptors explains Wolffian duct development in the Complete Androgen Insensitivity Syndrome. J Clin Endocrinol Metab 89: 5815.5822.
7. Melo KFS, Mendonca BB, Billerbeck AEC, et al, 2003 Clinical, hormonal, behavioral, and genetic characteristics of Androgen Insensitivity Syndrome in a Brazilian cohort: Five novel mutations in the androgen receptor gene. J Clin Endocrinol Metab 88: 3241.3250.
8. Adamopoulos D, Kapolla N, Nicopoulou S, Pappa A, Koukkou E, Gregoriou A, 2003 Assessment of Sertoli cell functional reserve and its relationship to sperm parameters. Int J Androl 26: 215-225.
9. Kubini K, Zachmann M, Albers N, et al, 2000 Basal inhibin B and the testosterone response to human chorionic gonadotropin correlate in prepubertal boys. J Clin Endocrinol Metab 85: 134.138.
10. Jockenhovel F, Rutgers JKL, Mason JS, Griffin JE, Swerdloff RS, 1993 Leydig cell neoplasia in a patient with Reifenstein syndrome. Exp Clin Endocrinol 101: 365-370.
11. Rey R, Mebarki F, Forest MG, et al, 1994 Anti-Mullerian Hormone in children with Androgen Insensitivity. J Clin Endocrinol Metab 79: 960-964.
12. Manuel M, Katayama KP, Jones Jr HW, 1976 The age of occurrence of gonadal tumors in intersex patients with a Y chromosome. Am J Obstet Gynecol 124: 293-300.
13. Hurt WG, Bodurtha JN, McCall JB, Ali MM, 1989 Seminoma in pubertal patient with androgen insensitivity syndrome. Am J Obstet Gynecol 161: 530-531.
14. Rutgers JL, Scully RE, 1991 The androgen insensitivity syndrome (testicular feminization): a clinicopathological study of 43 cases. Int J Gynecol Pathol 10: 126-144.
15. Augusto D, Leteurtre E, De La Taille A, Gosselin B, Leroy X, 2002 Calretinin: A valuable marker of normal and neoplastic Leydig cells of the testis. Appl Immunohistochem Mol Morph 10: 159-162.
16. Krichen Makni S, Mnif Hachicha L, Ellouze S, et al, 2005 Feminizing testicular syndrome with multiple hamartomas and bilateral paratesticular leiomyomas. Rev Med Interne 26: 980.983.
Address correspondence and requests for reprints to:
Dimitrios G. Goulis, Unit of Reproductive Endocrinology, First Department of Obstetrics and Gynecology,
Papageorgiou General Hospital, Ring Road, Nea Efkarpia, GR-546 03, Thessaloniki, Greece,
FAX: +30-2310-991.510, e-mail: dgg30@otenet.gr
Received 05-04-06, Revised 05-06-06, Accepted 20-06-06