1Fourth Pediatric Department, 2Clinic of Ophthalmology of Aristotle University of Thessaloniki, Thessaloniki, Greece, 3Endocrinology and Diabetes Research Unit, Schneider Children's Medical Center, Petah-Tikva, Israel
Laron-type dwarfism is an autosomal recessive disorder caused by deletions or mutations of the growth hormone receptor gene. It is characterized by high circulating levels of growth hormone (GH) and low levels of insulin-like growth factor I (IGF-I). Patients are refractory to both endogenous and exogenous GH, and present severe growth retardation and obesity. Therapy with recombinant human insulin-like growth factor-I (rhIGF-I) accelerates linear growth. We describe a 2-year old girl with Laron syndrome, who presented with postnatal growth failure and hypoglycaemic seizures. Her evaluation disclosed high GH values during a glucagon test (peak GH value 170 ng/ml) and very low IGF I value (0.1 ng/ml) with no rise following GH administration. The growth velocity improved considerably with the administration of IGF I. Molecular analysis showed a heterozygous mutation on exon 4 of the GH receptor gene, inherited from the mother, a rather puzzling finding considering the clinical findings in mother and infant. This case constitutes the first report of Laron syndrome from Greece.
Growth failure, growth hormone insensitivity, Laron syndrome
INTRODUCTION
A genetic form of dwarfism, having clinical and biochemical features of growth hormone deficiency (GHD) but abnormally high concentrations of immunoreactive serum growth hormone, was first reported in 3 Yemenite Jewish siblings in 19661. Within 2 years, 19 additional Israeli patients of Mid-Eastern Jewish ancestry were identified2 and several patients have been reported worldwide since then3,4. The majority of patients are traced to Semitic or Mediterranean origin. Further studies showed that the GH molecule was normal5 and that the GH receptor gene was defective6,7. This explained the unresponsiveness to endogenous and exogenous GH and the inability to generate IGF-I8. Initially named Laron-type dwarfism, this condition is currently known as Laron syndrome (LS), primary GH insensitivity (GHI), or primary GH resistance syndrome9. With the introduction of the PCR method, it was found that the molecular defects in patients with LS vary from exon deletions7 to mutations in the extracellular10, transmembrane11,12 or intracellular domain13 of the GH receptor as well as by post-receptor defects14. We here report the first patient with LS of Hellenic origin.
PATIENT REPORT
A 19-month old girl was admitted to the fourth Pediatric Dept of Aristotle University because of failure to thrive. According to the parents, both weight and length had remained unchanged since the age of 6 months. She was the product of a full-term, uneventful gestation and delivery. Her birth weight, birth length and head circumference were normal, 3340 g, 50 cm and 33.5 cm, respectively. She was breast fed for one month. She had neonatal jaundice, urinary tract infection at 2 ½ months and bronchiolitis at 4 months, as well as one episode of gastroenteritis at the age of 6 months. At the ages of 12 and 15 months, the infant experienced two episodes of seizures, attributed to hypoglycaemia. She has a 4-year old brother who is growing along the 50th percentile. Her parents are unrelated and of Hellenic origin. Her mother originates from Asia Minor and the father from Halkidiki, a region in Northern Greece (Figure 1 ). Maternal and paternal heights were 163 cm and 171.5 cm, respectively. Diabetes or other chronic illnesses were not reported in the family.
Physical examination revealed an extremely short infant with a body length of 66 cm, who weighed 7800g (well below the third percentile for both length and weight). By contrast, the head circumference was 50cm (97th percentile). An open fontanel, frontal bossing, blue sclerae, hypoplastic nasal bridge (Figure 2), small hands and feet (acromicria) and delayed dental development were noted. She could not walk and all other gross motor milestones were delayed. Furthermore, she had a high-pitched voice. Lungs, heart and abdomen (liver, spleen) examination revealed no pathologic findings. A wide spectrum of diseases related to growth failure and seizures were excluded, as demonstrated by the detailed laboratory findings listed in Table 1 . Karyotype analysis showed a normal female, (46,XX). Serum GH values prior to and following a glucagon provocation test (1 mg IM) were high, namely 20, 120, 150, 170, 165 ng/ml at 0, 30, 60, 90 and 120 minutes, respectively. An IGF-I generation test following rhGH administration (0.1 U/kg/24hrs for four consecutive days) showed no change in the serum IGF-I values, which were 0.10 ng/ml on the first and fifth day of the test. Serum IGF-BP3 was also low for her age: 344.2 ng/ml (normal >1200 ng/ml). Bone age at the chronological age of 19 months corresponded to 6 months. Retinal vascularization was less than expected at fundoscopy15. (Table 2 )
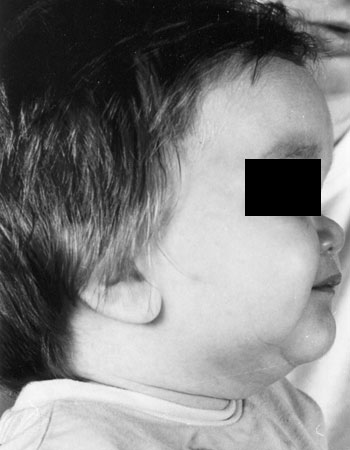
Figure 2. Typical Laron syndrome appearance of the patient, at the age of 19 months.
An MRI scan of the brain showed mild scaphocephaly, hypoplastic corpus callosum and lateral ventricular focal leucoencephalopathy, particularly in the frontal horns16. The hypothalamus and the pituitary gland were normal.
Molecular investigation of the GH receptor gene
DNA was isolated from peripheral blood leukocytes, both from the patient and her parents. All exons were amplified by PCR with specific intronic primers. Analysis of single strand conformation polymorphism (SSCP) was performed to detect aberrant bands. Direct sequencing of all exons disclosed that the patient carried a heterozygous mutation in exon 4 (R43X) and a heterozygous polymorphism in exon 6 (G168). The mutant allele was inherited from her heterozygous mother, who was homozygous for the polymorphism. The father was found to be heterozygous for the same polymorphism17.
Response to treatment
The patient was started on recombinant human insulin like growth factor I (rhIGF-I) at the dose of 100 ng/kg/day once a day subcutaneously. Frequent blood glucose measurements were performed on a portable glucometer, and a reduction in hypoglycemic episodes was registered. She started walking 2 months following the institution of therapy and her speech and dentition improved. In nine months she grew 8.4 cm and gained 12.5 kg in weight.
DISCUSSION
The diagnosis of Laron syndrome (LS) in this patient was based on the characteristic clinical features, the high serum values of growth hormone and the low IGF-I serum levels before and after GH administration (IGF-I generation test)1-3,8. The patient was found to have a heterozygous mutation on Exon 4 (R43X) and a heterozygous polymorphism on exon 6 (Gly168Gly). The mutation R43X was inherited from her mother, who is also heterozygous for this mutation. The mother is, in addition, homozygous for the polymorphism G168. The father was found to be heterozygous for the same polymorphism17. There is no explanation for the observation that the patient presents the complete typical phenotype of LS, whereas the mother, having the same defect, has normal height and normal features. From previous studies we know that heterozygous family members, carriers of molecular defects of the GH-R gene, are of normal height, with the exception of a few mothers18,19. The father, who is heterozygous for the polymorphism, is not expected to have a pathologic phenotype.
The origin of most patients with LS reported previously is in the Mediterranean region, the Middle East and the Indian subcontinent3,20,21. Consanguinity was common in most of the above populations. This is the first patient of LS of Hellenic origin. The patient, however, traces her origin to Asia Minor (great-grandparents). There is no consanguinity between her parents and none reported to have accrued in previous generations. The mutation of GHR on Exon 4 detected in this patient is one of 41 reported to date and has been found in patients originating from the Mediterranean region19.
The brain abnormalities obtained by MRI were attributed to the lack of IGF-I, which is known to play a significant role in the development of the brain and bony structures of the cranium16.
Treatment with rhIGF-I improved height velocity, body weight and serum IGF-I levels in this patient. The rhIGF-1 therapy is being well tolerated except for some hypoglycemic episodes due to irregular feeding habits. The parents were instructed to recognize the signs and symptoms of hypoglycemia, and to monitor blood glucose at home. Her psychomotor development, extremely delayed at diagnosis, improved significantly following the introduction of therapy. Her final intellectual development cannot be predicted. Large variability, ranging from normal intelligence to severe mental retardation has been reported22. Despite the difficulties in the current availability of the drug, it is hoped that it will be possible to treat the patient continuously to prevent the late consequences of lack of therapy23.
REFERENCES
1. Laron Z, Pertzelan A, Mannheimer S, 1966 Genetic pituitary dwarfism with high serum concentration of growth hormone—a new inborn error of metabolism? Isr J Med Sci 2: 152-155.
2. Laron Z, Pertzelan A, Karp M, 1968 Pituitary dwarfism with high serum levels of growth hormone. Isr J Med Sci 4: 883-894.
3. Laron Z, 1999 Natural history of the classical form of primary growth hormone (GH) resistance (Laron syndrome). J Pediatr Endocrinol Metab 12: 231-249.
4. Rosenfeld RG, Rosenbloom AL, Guevara-Aguirre J, 1994 Growth hormone (GH) insensitivity due to primary GH receptor deficiency. Endocr Rev 15: 369-390.
5. Eshet R, Peleg S, Josefsberg Z, Fuchs C, Arnon R, Laron Z, 1985 Some properties of the plasma hGH activity in patients with Laron-type dwarfism determined by a radioreceptor assay using human liver tissue. Horm Res 22: 276-283.
6. Eshet R, Laron Z, Pertzelan A, Dintzman M, 1984 Defect of human growth hormone receptors in the liver of two patients with Laron-type dwarfism. Isr J Med Sci 20: 8-11.
7. Godowski PJ, Leung DW, Meacham LR, et al, 1989 Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc Natl Acad Sci USA 86: 8083-8087.
8. Laron Z, Pertzelan A, Karp M, Kowadlo-Silbergeld A, Daughaday WH , 1971 Administration of growth hormone to patients with familial dwarfism with high plasma immunoreactive growth hormone. Measurement of sulfation factor, metabolic, and linear growth responses. J Clin Endocrinol Metab 33: 332-342.
9. Laron Z, Blum W, Chatelain P, et al, 1993 Classification of growth hormone insensitivity syndrome. J Pediatr 122: 241.
10. Amselem S, Duquesnoy P, Attree O, et al, 1989 Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med 321: 989-995.
11. Silbergeld A, Dastot F, Klinger B, et al, 1997 Intronic mutation in the growth hormone (GH) receptor gene from a girl with Laron syndrome and extremely high serum GH binding protein: extended phenotype study in a very large pedigree. J Pediatr Endocrinol Metab10: 265-274.
12. Ayling RM, Ross R, Towner P, et al, 1997 A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nature Genet 16:13-14.
13. Woods KA, Fraser NN, Postel-Vinay MC, Savage MO, Clark AJL, 1996 A homozygous splice site mutation affecting the intracellular domain of the growth (GH) receptor resulting in Laron syndrome with elevated GH-binding protein. J Clin Endocrinol Metab 81: 1686-1690.
14. Laron Z, Klinger B, Eshet R, Kaneti H, Karasik A, Silbergeld A, 1993 Laron syndrome due to a post-receptor defect: response to IGF-1 treatment. Isr J Med Sci 29: 757-763.
15. Hellerstrom A, Carlsson B, Niklasson A, et al, 2002 IGF-I is critical for normal vascularization of the human retina. J Clin Endocrinol Metab 87: 3413-3416.
16. Kornreich L, Horev G, Schwarz M, Karmazyn B, Laron Z, 2002 Craniofacial and brain abnormalities in Laron syndrome (primary growth hormone insensitivity). Eur J Endocrinol 146: 499-503.
17. Galli_Tsinopoulou A, Shevah O, Laron Z, 2003 Classical phenotype of Laron Syndrome in a girl with a heterozygous mutation and a heterozygous polymorphism of the GH receptor gene. J Pediat Endocrinol Metab (in press).
18. Laron Z, Klinger B, Erster B, Silbergeld A, 1989 Serum
GH binding protein activity identifies the heterozygous carriers for Laron type dwarfism. Acta Endocrinol 121: 603-608.
19. Amselem S, Sobrier ML, Duquesnoy P, et al, 1991 Recurrent nonsense mutations in the growth hormone receptor from patients with Laron dwarfism. J Clin Invest 87: 1098-1102.
20. Rosenbloom AL, 1999 Growth hormone insensitivity: physiologic and genetic basis, phenotype, and treatment. J Pediatr 135: 280-289. 21. Laron Z 2002 Growth hormone resistance (Laron syndrome) In: Chrousos G (ed) Hormone Resistance and Hypersensitivity States, Lippincott Williams & Wilkins, New York; pp, 251-267.
22. Galatzer A, Aran O, Nagelberg N, Rubitzek J, Laron Z 1993 Congenital and psychosocial functioning of young adults with Laron Syndrome In: Laron Z, Parks JS (eds) Lessons from Laron Syndrome (LS) 1966-1992, Pediatr Adolesc Endocrinol, Basel, Karger; pp, 53-60.
23. Laron Z, 2001 Consequences of not treating children with Laron Syndrome (primary growth hormone insensitivity) J Pediatr Endocrinol Metab 14: 1243-1248.
Address correspondence and requests for reprints to:
Assimina Galli-Tsinopoulou, MD, 8 Hydras str., 552 36
Thessaloniki, Greece, Tel.: +30 2310 342 374,+30 2310 994 826,
Fax: +30 2310 331263, e-mail: galtsin@otenet.gr - assimina@med.auth.gr
Received 15-01-03, Revised 28-02-03, Accepted 07-03-03