1Ph.D., Department of Pharmacy, 2M.D., Department of Internal Medicine, 3M.D., Department of Obstetrics and Gynecology, Medical College of Virginia Campus, Virginia Commonwealth University, Richmond, Virginia
Supported in part by NIH grants to J.E.N.: RO1HD35629 and K24HD40237
Inositolphosphoglycans, D-chiro-inositol, Myo-inositol, Polycystic ovary syndrome, Insulin resistance
Current evidence suggests that insulin resistance contributes to the pathophysiologic features of the polycystic ovary syndrome (PCOS). The observation that insulin resistance is a central feature of PCOS leads to studies of the insulin signaling system in this disorder. This article reviews the possible role of putative insulin mediators, inositolphosphoglycans (IPG) mediators, in PCOS. We will focus on how IPGs mediate (a) hyperandrogenism and (b) insulin sensitivity in this disorder.
INSULIN RESISTANCE AND THE PATHOGENESIS OF PCOS
PCOS affects approximately 6-10% of women of reproductive age, and is characterized by chronic anovulation and hyperandrogenism. Insulin resistance, with its compensatory hyperinsulinemia, is a virtually
universal feature of PCOS1-3. There is a form of insulin resistance that is intrinsic to the disorder and is present even in lean women with PCOS3. In addition, women with PCOS have an added component of insulin resistance from adiposity. About 50% to 80% of women with PCOS are obese, and obesity itself induces insulin resistance and hyperinsulinemia4.
Insulin resistance, along with its concomitant hyperinsulinemia, is critical in the pathogenesis of hyperandrogenism in PCOS. In vitro evidence shows that in isolated thecal cells from PCOS women, insulin stimulates ovarian testosterone production5. In vivo evidence also supports the role of hyperinsulinemia in the elevated circulating levels of testosterone in PCOS. Suppression of insulin release by diazoxide decreases both serum total and free testosterone levels in women with PCOS. In addition, when insulin-sensitizing agents such as metformin6-8, troglitazone9,10, and more recently, rosiglitazone11,12, are administered to women with PCOS, serum testosterone concentrations decrease together with serum insulin concentrations. Besides playing a role in the anovulatory infertility of PCOS, insulin resistance in PCOS is associated with an increased risk of hypertension13,14, dyslipidemia15-18, type 2 diabetes19-23, metabolic syndrome24, and cardiovascular disease25-29.
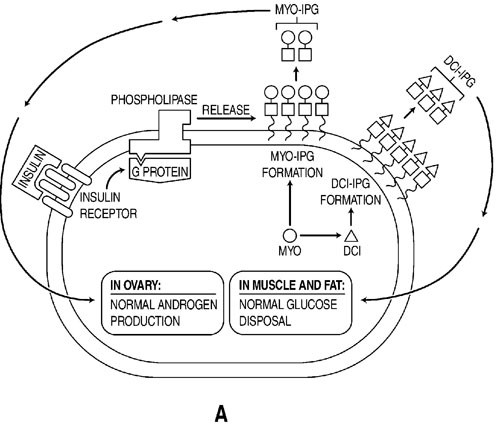
The above observations suggest that the hyperinsulinemia of PCOS plays an important role in the hyperandrogenism of the disorder. However, these observations also suggest a contradictory tenet to the insulin resistance hypothesis in PCOS: while PCOS women are "insulin resistant" in terms of glucose disposal, their ovarian tissues remain sensitive to insulin's stimulation in terms of testosterone biosynthesis. In an attempt to provide a unifying hypothesis for these phenomena, we propose that ovarian androgen biosynthesis and glucose homeostasis in PCOS may be mediated via a signaling system for insulin that is separate from the classical tyrosine phosphate signaling mechanism.
THE INOSITOLPHOSPHOGLYCAN SIGNALING SYSTEM
Some actions of insulin may involve low molecular weight inositolphosphoglycan (IPG) mediators, also known as putative insulin mediators or second messengers30-32. When insulin associates with its receptor, mediators of this class are generated by hydrolysis of glycosylphosphatidylinositol lipids and/or proteinated species located at the outer leaflet of the cell membrane. Released mediators are then internalized and alter the activities of purified enzymes33,34 and the metabolism of intact cells32,35 in an insulin mimetic fashion. Two different IPG mediators have been extracted from rat liver36. One of these is the D-chiro-inositolphosphoglycan mediator (DCI-IPG) which activates pyruvate dehydrogenase (PDH) phosphatase. The other IPG mediator is the myo-inositolphosphoglycan mediator (MYO-IPG) which inhibits cyclic AMP-dependent protein kinase (PKA). These IPG mediators may mediate the alternate insulin signaling mechanism.
IPGS AND OVARIAN ANDROGEN BIOSYNTHESIS IN PCOS
Over a decade ago, experiments from our laboratory examined whether IPGs mediate insulin's steroidogenic actions in human placental tissue. In studies using placental cytotrophoblasts isolated from human placentae, purified liver extracts containing either the MYO-IPG or the DCI-IPG mimicked insulin's inhibitory action on aromatase37 and stimulatory action on 3â-hydroxysteroid dehydrogenase38 in a concentration dependent fashion39. In this system, it was also shown that MYO-IPG may be more potent than DCI-IPG39. Preincubation of the cytotrophoblasts with a polyclonal antibody that blocked IPGs completely abolished insulin's ability to either inhibit aromatase or stimulate 3b-hydroxysteroid dehydrogenase activity. These studies suggested that the IPG system may be responsible for transducing insulin's regulation of human placental steroidogenesis.
Similarly, Romero et al. demonstrated that IPG mediators mimicked insulin's stimulation of progesterone production by swine granulosa cells40. In addition, insulin's action on progesterone synthesis was abolished when the swine cells were preincubated with an antibody that blocked the IPG mediators40. These observations suggested that IPGs mediate insulin's steroidogenic actions in swine granulosa cells.
In order to assess specifically in PCOS the mediation of insulin stimulated steroidogenic action by IPGs, our laboratory and collaborators in Caracas, Venezuela, obtained ovarian tissue from PCOS women undergoing gynecologic surgery41. Thecal cells, the cells responsible for ovarian testosterone biosynthesis, were isolated and cultured. Upon exposure to insulin, testosterone biosynthesis increased 10- to 12- fold in these primary thecal cultures. Experiments were also performed using a putative chiro-IPG mediator, named "INS-2" (provided by Insmed Pharmaceuticals). INS-2 is a synthetic pseudo-disaccharide of pinitol (3-O-methyl D-chiro-inositol) and galactosamine that is believed to be closely related to the pH 2.0 DCI-IPG putative insulin mediator described by Larner and colleagues41,43. Exposure to the INS-2 mediator led to thecal testosterone biosynthesis in a dose-dependent fashion. In addition, the INS-2 mediator in micromolar concentrations stimulated thecal testosterone biosynthesis to a degree at least equal to and sometimes greater than that of insulin in maximal concentrations. These observations suggested that the synthetic INS-2 mediator could mimic insulin's action in stimulating thecal testosterone biosynthesis.
To confirm that insulin stimulated testosterone biosynthesis via the IPG signal transduction system, PCOS thecal cells were preincubated with the polyclonal anti-inositol antibody A23939. Preincubation with A23939 completely inhibited insulin's ability to stimulate testosterone biosynthesis. These results were confirmed by using a second anti-inositolglycan antibody, aIPG, that had been shown to block the action of inositolglycan mediators44. Preincubation with aIPG similarly abolished the ability of insulin to stimulate thecal testosterone biosynthesis. The inhibitory effect of the anti-inositol antibodies was specific for insulin, since preincubation with these blocking antibodies did not affect stimulation of testosterone biosynthesis by hCG.
The results of these studies demonstrated that a synthetic DCI-IPG (INS-2) mimicked insulin's stimulation of thecal testosterone biosynthesis in a dose-responsive manner and to a degree at least equal to that of insulin. These studies also suggested that IPG mediators may serve as the signal transduction system for insulin's stimulation of human thecal testosterone biosynthesis. It is important to note, however, that these studies were not designed to identify which of the many IPGs was primarily responsible for mediating insulin's action on steroidogenesis, but as reported previously, the MYO-IPG was more potent in human placental steroidogenesis than DCI-IPG.
IMPROVEMENT OF INSULIN SENSITIVITY IN PCOS BY D-CHIRO-INOSITOL
Besides IPGs' role in transducing androgen production in PCOS, available data suggest that the IPG system also mediates glucose metabolism. Among the different species of IPG mediators that have been identified, several lines of evidence support the hypothesis that a deficiency specifically of DCI-IPG may result in insulin resistance. Although several studies have linked insulin resistance (as determined by the hyperinsulinemic-euglycemic clamp technique) with decreased urinary excretion of total DCI45-48, large urinary losses of DCI have been shown in both type 1 and type 2 diabetes using a more sensitive gas chromatography/mass-spectroscopy technology49. This urinary loss of DCI was out of proportion to that expected in diabetic glycosuria. Furthermore, significantly decreased muscle DCI-IPG bioactivity and decreased total DCI content have been noted in needle biopsies48 and autopsy specimens50 from type 2 diabetic subjects compared with controls. In a study of rats, intragastric administration of DCI significantly decreased hyperglycemia in low-dose streptozocin rats and improved glucose tolerance in normal rats51. Administered intravenously to monkeys with varying degrees of insulin resistance, DCI accelerated glucose disposal and increased the rate of decrease of plasma insulin51. These observations suggested that a deficiency in DCI-IPG may lead to insulin resistance, and that exogenous administration of DCI may enhance insulin sensitivity and improve insulin action in insulin-resistant individuals.
To determine if DCI-IPG is deficient specifically in women with PCOS, a preliminary study was conducted to compare the total DCI content in urine and blood of women with PCOS to that of age- and weight-matched normal women, and to determine if this difference may be accompanied by (a) decreased release of DCI-IPG (as determined by bioactivity) in blood of women with PCOS during an oral glucose challenge and (b) decreased whole-body insulin sensitivity52.
As reported in preliminary form, urinary DCI clearance was 5-fold higher in PCOS as compared with normal women (p=0.006). Urinary DCI clearance was also correlated inversely with insulin sensitivity as defined by the Bergman minimal model (p=0.0003). Furthermore, DCI-IPG release was impaired in response to insulin release in women with PCOS upon oral glucose challenge, as shown by a 3-fold AUCDCI-IPG/AUCInsulin decrease in PCOS (p<0.0001)52.
These results confirmed a functional deficiency of DCI in women with PCOS, and suggested that DCI deficiency may be related to increased urinary DCI clearance and may contribute to insulin resistance of the syndrome. These data also corroborated a previous study showing that urinary DCI excretion was increased in insulin resistant states49. However, these data did not rule out the existence of either an intra-tissue deficiency of DCI, or a defect in incorporating DCI into the DCI-IPG mediator.
If a functional deficiency of this specific IPG mediator, DCI-IPG, is responsible for insulin resistance in PCOS, then administration of DCI might replenish DCI-IPG stores and improve insulin sensitivity. To test this hypothesis, 44 obese women (BMI >28 kg/m2) with PCOS were given DCI 1200 mg orally or placebo once daily for six to eight weeks53. Fasting serum steroids were measured and oral glucose tolerance tests were performed before and after the treatment period. Serum progesterone was obtained weekly to monitor for ovulation.
In the women given DCI (n=22), the mean AUCinsulin (±SD) curve after oral glucose challenge decreased from 13417±2467 to 5158±1431 µU/ml/min (P= 0.007)53. In women who demonstrated impaired glucose tolerance at baseline, AUCglucose during the OGTT decreased from 13983±3625 to 10945±2410 mg/dl/min (P=0.03), resulting in normal glucose tolerance curves in the six women given DCI, but no change in the women given placebo. Diastolic (P<0.001) and systolic (P=0.05) blood pressures decreased by 4 mmHg, and plasma triglyceride concentrations decreased from 184±88 to 110±61 mg/dl (P=0.002 compared to the placebo group). These parameters did not change substantially in the placebo group. Moreover, ovulation occurred in 19 of the 22 women (86%) who received DCI, but only in 6 of the 22 women (27%) in the placebo group (P<0.001). Serum free testosterone in all women treated with DCI decreased by 53% from 1.1±0.8 to 0.5±0.5 ng/dl and did not change substantially in the placebo group (p=0.006 for the comparison).
The above study showed that administration of DCI enhanced insulin action in PCOS, thereby improving ovulation and decreasing serum androgen concentrations. DCI administration in PCOS also ameliorated components of the metabolic syndrome. These results support the idea that the insulin resistance of PCOS is, at least in part, related to a functional deficiency in DCI, and presumably DCI-IPG.
To further test the hypothesis that DCI deficiency contributes to the insulin resistance of PCOS independent of adiposity, a study similar to the above investigation was conducted in lean women with PCOS to determine if DCI administration was beneficial in this subset54. In this study, 600 mg DCI or placebo once daily was administered for six to eight weeks. As in the previous study, DCI administration improved insulin sensitivity, ovulation and serum androgen levels in these lean women with PCOS. These findings confirmed that lean women with PCOS also suffered from insulin resistance and a functional deficiency in DCI may contribute to their insulin resistance.
Since DCI deficiency seems to contribute to insulin resistance in PCOS, we wondered whether some of the beneficial effects of insulin sensitizing drugs in this disorder may be mediated by improvements in DCI-IPG release. We utilized stored serum samples from a study where we administered metformin or placebo for 4 to 8 weeks, and quantified DCI-IPG release from these stored samples55. Nineteen obese
(BMI ³ 27.5 kg/m2) women with PCOS who took metformin (n=10) or placebo (n=9) were investigated. Insulin and DCI-IPG release during an oral glucose tolerance test were assessed. Treatment with metformin decreased the AUCinsulin (±SE) compared to placebo during oral glucose challenge (-3574 ± 962 vs. +1367 ± 1021 mIU/min x ml, p=0.003). In addition, after metformin therapy, the bioactive DCI-IPG per unit of insulin (AUCDCI-IPG/AUCinsulin) increased by 160%. In comparison, this value decreased by 29% after placebo (p=0.002). This study suggested the possibility that metformin improved insulin sensitivity in PCOS in part by improving insulin-stimulated release of DCI-IPG mediators.
In summary, these findings support the idea that DCI deficiency contributes to insulin resistance in PCOS. They further suggest that DCI deficiency in PCOS is unrelated to adiposity, since it appears to be present in both obese and lean women with PCOS. In addition, exogenous DCI administration in lean and obese women with PCOS can overcome DCI deficiency and alleviate insulin resistance in the disorder. Finally, some of the improvement in insulin sensitivity mediated by metformin may be related to an improvement in insulin-stimulated release of DCI-IPG mediators.
A UNIFYING HYPOTHESIS: THE ROLE OF IPGS IN BOTH GLUCOSE HOMEOSTASIS AND HYPERANDROGENEMIA IN PCOS
The above findings present us with an apparent paradox. If IPG mediators are responsible in part for mediating insulin sensitivity and also transducing ovarian andgrogen production, then one might expect that a deficiency of IPG mediators in PCOS would logically lead to insulin resistance and a low level of circulating androgens. DCI administration would therefore increase circulating androgens in women with PCOS. Yet this is not the case since DCI administration to women with PCOS improves insulin sensitivity, improves ovulation and lowers circulating androgens53,54. How does the IPG transduction system explain both the insulin resistance and the hyperandrogenism of this disorder?
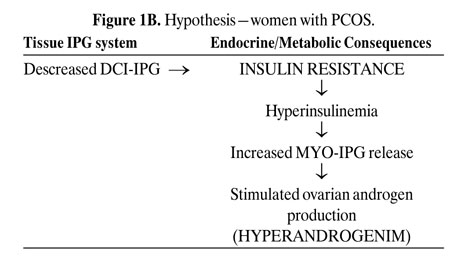
In this regard, it is important to emphasize that while studies have shown that DCI-IPG deficiency in PCOS may contribute to the insulin resistance of the syndrome, studies in steroidogenesis have not identified which of the many IPGs are responsible for transducing insulin's signal in vivo for testosterone biosynthesis. In human placental cytotrophoblasts in vitro, MYO containing IPG seems to be more potent than DCI-IPG in mediating steroidogenesis39. We postulate that MYO-IPG, but not DCI-IPG, may be the primary mediator for testosterone biosynthesis in PCOS, although this theory awaits validation by further studies. In contrast, evidence suggests that DCI-IPG mediates insulin sensitivity in PCOS.
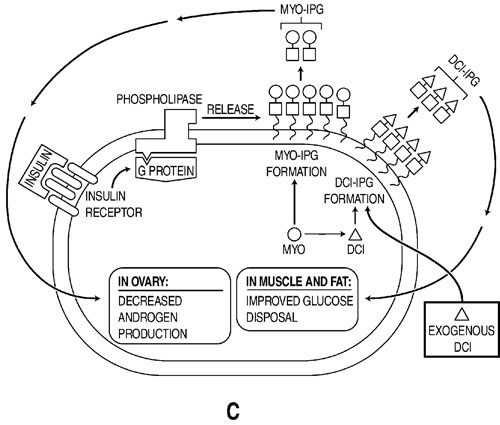
Figures 1 A-C depict our hypothesis. Levels of DCI-IPG and MYO-IPG are normal in the absence of PCOS (Figure 1A). In PCOS, a functional deficiency in DCI-IPG exists as supported by available data, and we hypothesize that there may be an increased level of MYO-IPG due to compensatory hyperinsulinemia resulting from insulin resistance (Figure 1B). Administration of DCI restores normal levels of DCI-IPG, and may improve insulin sensitivity. Decreased circulating insulin then results in decreased MYO-IPG release, reducing ovarian testosterone biosynthesis (Figure 1C). To date, we have preliminary data supporting a functional deficiency of DCI in PCOS52. Additional studies are underway to assess whether there is an excess of MYO-IPG in this disorder.



CONCLUSION AND FUTURE DIRECTIONS
IPGs play a role in both ovarian testosterone biosynthesis and insulin resistance in PCOS. Deficiency of DCI-IPG may be in part responsible for the insulin resistance in PCOS. DCI administration ameliorates insulin resistance, hyperandrogenemia and improves ovulation in this disorder. We postulate that MYO-IPG may be the primary IPG responsible for ovarian testosterone biosynthesis, but this awaits validation by future studies. Additional studies should also investigate the ratio of DCI-IPG to MYO-IPG in ovarian tissues in PCOS women. Therapeutic moieties to decrease MYO-IPG or increase DCI-IPG may be a possible research avenue to correct the hyperandrogenism and insulin resistance of this disorder.
REFERENCES
1. Dunaif A, Graf M, Mandeli J, Laumas V, Dobrjansky A, 1987 Characterization of groups of hyperandrogenic women with acanthosis nigricans, impaired glucose tolerance, and/or hyperinsulinemia. J Clin Endocrinol Metab 65: 499-507.
2. Dunaif A, Mandeli J, Fluhr H, Dobrjansky A, 1988 The impact of obesity and chronic hyperinsulinemia on gonadotropin release and gonadal steroid secretion in the polycystic ovary syndrome. J Clin Endocrinol Metab 66: 131-139.
3. Dunaif A, Segal KR, Futterweit W, Dobrjansky A, 1989 Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 38: 1165-1174.
4. Campbell PJ, Gerich JE, 1990 Impact of obesity on insulin action in volunteers with normal glucose tolerance: demonstration of a threshold for the adverse effect of obesity. J Clin Endocrinol Metab 70: 1114-1118.
5. Nestler JE, Jakubowicz DJ, Falcon de Vargasm A, Brik C, Quinterro N, Medina F, 1998 Insulin stimulates testosterone biosynthesis by huan thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 83: 2001-2005.
6. Lord JM, Flight IHK, Norman RJ, 2003 Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ 327: 951-960.
7. Nestler JE, Jakubowicz DJ, 1996 Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N Engl J Med 335: 617-623.
8. Nestler JE, Jakubowicz DJ, 1997 Lean women with polycystic ovary syndrome respond to insulin reduction with decreases in ovarian P450c17{alpha} activity and serum androgens. J Clin Endocrinol Metab 82: 4075-4079.
9. Dunaif A, Scott D, Finegood D, Quintana B, Whitcomb R, 1996 The insulin-sensitizing agent troglitazone improves metabolic and reproductive abnormalities in the polycystic ovary syndrome. J Clin Endocrinol Metab 81: 3299-3306.
10. Ehrmann DA, Schneider DJ, Sobel BE, et al, 1997 Troglitazone improves defects in insulin action, insulin secretion, ovarian steroidogenesis, and fibrinolysis in women with polycystic ovary syndrome. J Clin Endocrinol Metab 82: 2108-2116.
11. Ghazeeri G, Kutteh WH, Bryer-Ash M, Haas D, Ke RW, 2003 Effect of rosiglitazone on spontaneous and clomiphene citrate-induced ovulation in women with polycystic ovary syndrome. Fertil Steril 79: 562-566.
12. Shobokshi A, Shaarawy M, 2003 Correction of insulin resistance and hyperandrogenism in polycystic ovary syndrome by combined rosiglitazone and clomiphene citrate therapy. J Soc Gynecol Investig 10: 99-104.
13. Bjorntorp P, 1996 The android woman—a risky condition. J Intern Med 239: 105-110.
14. Rebuffe-Scrive M, Cullberg G, Lundberg PA, Lindstedt G, Bjorntorp P, 1989 Anthropometric variables and metabolism in polycystic ovarian disease. Horm Metab Res 21: 391-397.
15. Conway GS, Agrawal R, Betteridge DJ, Jacobs HS, 1992 Risk factors for coronary artery disease in lean and obese women with the polycystic ovary syndrome. Clin Endocrinol 37: 119-125.
16. Legro RS, Kunselman AR, Dunaif A, 2001 Prevalence and predictors of dyslipidemia in women with polycystic ovary syndrome. Am J Med 111: 607-613.
17. Robinson S, Henderson AD, Gelding SV, et al, 1996 Dyslipidaemia is associated with insulin resistance in women with polycystic ovaries. Clin Endocrinol 44: 277-284.
18. Talbott E, Clerici A, Berga SL, et al, 1998 Adverse lipid and coronary heart disease risk profiles in young women with polycystic ovary syndrome: results of a case-control study. J Clin Epidemiol 51: 415-422.
19. Dahlgren E, Johansson S, Lindstedt G, et al, 1992 Women with polycystic ovary syndrome wedge resected in 1956 to 1965: a long-term follow-up focusing on natural history and circulating hormones. Fertil Steril 57: 505-513.
20. Dunaif A, Graf M, Mandeli J, Laumas V, Dobrjansky A, 1987 Characterization of groups of hyperandrogenic women with acanthosis nigricans, impaired glucose tolerance, and/or hyperinsulinemia. J Clin Endocrinol Metab 65: 499-507.
21. Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J, 1999 Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care 22: 141-146.
22. Legro RS, Kunselman AR, Dodson WC, Dunaif A, 1999 Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab 84: 165-169.
23. Wild S, Pierpoint T, McKeigue P, Jacobs H, 2000 Cardiovascular disease in women with polycystic ovary syndrome at long-term follow-up: a retrospective cohort study. Clin Endocrinol 52: 595-600.
24. Taponen S, Martikainen H, Jarvelin MR, et al, 2004 Metabolic cardiovascular disease risk factors in women with self-reported symptoms of oligomenorrhea and/or hirsutism: Northern Finland Birth Cohort 1966 Study. J Clin Endocrinol Metab 89: 2114-2118.
25. Christian RC, Dumesic DA, Behrenbeck T, Oberg AL, Sheedy PF, Fitzpatrick LA, 2003 Prevalence and predictors of coronary artery calcification in women with polycystic ovary syndrome. J Clin Endocrinol Metab 88: 2562-2568.
26. Dahlgren E, Janson PO, Johansson S, Lapidus L, Oden A, 1992 Polycystic ovary syndrome and risk for myocardial infarction. Evaluated from a risk factor model based on a prospective population study of women. Acta Obstet Gynecol Scand 71: 599-604.
27. Legro RS, 2003 Polycystic ovary syndrome and cardiovascular disease: a premature association? Endocr Rev 24: 302-312.
28. Paradisi G, Steinberg HO, Hempfling A, et al, 2001 Polycystic ovary syndrome is associated with endothelial dysfunction. Circulation 103: 1410-1415.
29. Talbott EO, Guzick DS, Sutton-Tyrrell K, et al, 2000 Evidence for association between polycystic ovary syndrome and premature carotid atherosclerosis in middle-aged women. Arterioscler Thromb Vasc Biol 20: 2414-2421.
30. Larner J, 1983 Mediators of postreceptor action of insulin. Am J Med 74: 38-51.
31. Larner J, 1988 Insulin-signaling mechanisms. Lessons from the old testament of glycogen metabolism and the new testament of molecular biology. Diabetes 37: 262-275.
32. Saltiel AR, 1990 Second messengers of insulin action. Diabetes Care 13: 244-256.
33. Jarett L, Seals JR, 1979 Pyruvate dehydrogenase activation in adipocyte mitochondria by an insulin-generated mediator from muscle. Science 206: 1407-1408.
34. Larner J, Galasko G, Cheng K, et al, 1979 Generation by insulin of a chemical mediator that controls protein phosphorylation and dephosphorylation. Science 206: 1408-1410.
35. Larner J, Romero G, Kennington AS, Lilley K, Kilgour E, Zhang C et al., 1990 Duality in the mechanism of action of insulin. Adv Second Messenger Phosphoprotein Res 24: 290-294.
36. Larner J, Huang LC, Schwartz CF, et al, 1988 Rat liver insulin mediator which stimulates pyruvate dehydrogenase phosphate contains galactosamine and D-chiroinositol. Biochem Biophys Res Commun 151: 1416-1426.
37. Nestler JE, 1987 Modulation of aromatase and P450 cholesterol side-chain cleavage enzyme activities of human placental cytotrophoblasts by insulin and insulin-like growth factor I. Endocrinology 121: 1845-1852.
38. Nestler JE, 1989 Insulin and insulin-like growth factor-I stimulate the 3 beta-hydroxysteroid dehydrogenase activity of human placental cytotrophoblasts. Endocrinology 125: 2127-2133.
39. Nestler JE, Romero G, Huang LC, Zhang CG, Larner J, 1991 Insulin mediators are the signal transduction system responsible for insulin's actions on human placental steroidogenesis. Endocrinology 129: 2951-2956.
40. Romero G, Garmey JC, Veldhuis JD, 1993 The involvement of inositol phosphoglycan mediators in the modulation of steroidogenesis by insulin and insulin-like growth factor-I. Endocrinology 132: 1561-1568.
41. Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F, 1998 Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 83: 2001-2005.
42. Larner J, Price JD, Heimark D, et al, 2003 Isolation, structure, synthesis, and bioactivity of a novel putative insulin mediator. A galactosamine chiro-inositol pseudo-disaccharide Mn2+ chelate with insulin-like activity. J Med Chem 46: 3283-3291.
43. Larner J, Allan G, Sleevi M, Price J, Rule G, Piccariello T, 1997 Molecular mechanisms of insulin resistance. Structure and formation of a novel pinitol-galactosamine disaccharide—a putative insulin mediator. Program of the 16th International Diabetes Federation Congress 1997.
44. Romero G, Gamez G, Huang LC, Lilley K, Luttrell L, 1990 Anti-inositolglycan antibodies selectively block some of the actions of insulin in intact BC3H1 cells. Proc Natl Acad Sci USA 87: 1476-1480.
45. Ortmeyer HK, Bodkin NL, Lilley K, Larner J, Hansen BC, 1993 Chiroinositol deficiency and insulin resistance. I. Urinary excretion rate of chiroinositol is directly associated with insulin resistance in spontaneously diabetic rhesus monkeys. Endocrinology 132: 640-645.
46. Suzuki S, Kawasaki H, Satoh Y, et al, 1994 Urinary chiro-inositol excretion is an index marker of insulin sensitivity in Japanese type II diabetes. Diabetes Care 17: 1465-1468.
47. Craig JW, Larner J, Asplin CM, Chiro-inositol deficiency and insulin resistance. In: Draznin B, LeRoith D, editors. Molecular Biology of Diabetes II. Totowa, N.J.: Humana Press, 1998: 343-362.
48. Kennington AS, Hill CR, Craig J, et al, 1990 Low urinary chiro-inositol excretion in non-insulin-dependent diabetes mellitus. N Engl J Med 323: 373-378.
49. Ostlund RE Jr, McGill JB, Herskowitz I, Kipnis DM, Santiago JV, Sherman WR, 1993 D-chiro-inositol metabolism in diabetes mellitus. Proc Natl Acad Sci USA 90: 9988-9992.
50. Asplin I, Galasko G, Larner J, 1993 chiro-inositol deficiency and insulin resistance: a comparison of the chiro-inositol- and the myo-inositol-containing insulin mediators isolated from urine, hemodialysate, and muscle of control and type II diabetic subjects. Proc Natl Acad Sci USA 90: 5924-5928.
51. Ortmeyer HK, Huang LC, Zhang L, Hansen BC, Larner J, 1993 Chiroinositol deficiency and insulin resistance. II. Acute effects of D-chiroinositol administration in streptozotocin-diabetic rats, normal rats given a glucose load, and spontaneously insulin-resistant rhesus monkeys. Endocrinology 132: 646-651.
52. Baillargeon J-P, Diamanti-Kandarakis E, Apridonidze T, Iuorno MJ, Nestler JE. 2004 D-chiro-inositol clearance, insulin stimulated release of D-chiro-inositol-containing inositolphosphoglycan and insulin sensitivity in women with polycystic ovary syndrome. The Endocrine Society 86th Annual Meeting, New Orleans, Louisiana OR31-2, 118.
53. Nestler JE, Jakubowicz DJ, Reamer P, Gunn RD, Allan G, 1999 Ovulatory and metabolic effects of D-chiro-inos
itol in the polycystic ovary syndrome. N Engl J Med 340: 1314-1320.
54. Iuorno MJ, Jakubowicz DJ, Baillargeon JP, et al, 2002 Effects of D-chiro-inositol in lean women with the polycystic ovary syndrome. Endocr Pract 8: 417-423.
55. Baillargeon JP, Iuorno MJ, Jakubowicz DJ, Apridonidze T, He N, Nestler JE, 2004 Metformin therapy increases insulin-stimulated release of D-chiro-inositol-containing inositolphosphoglycan mediator in women with polycystic ovary syndrome. J Clin Endocrinol Metab 89: 242-249.
Address correspondence and requests for reprints to:
John E. Nestler, M.D., Virginia Commonwealth University,
MCV Campus, PO Box 980111, Richmond, VA 23298-0111, e-mail: nestler@hsc.vcu.edu
Received 17-08-04, Revised 20-09-04, Accepted 25-09-04